TCF2 Attenuates FFA-Induced Damage in Islet β-Cells by Regulating Production of Insulin and ROS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
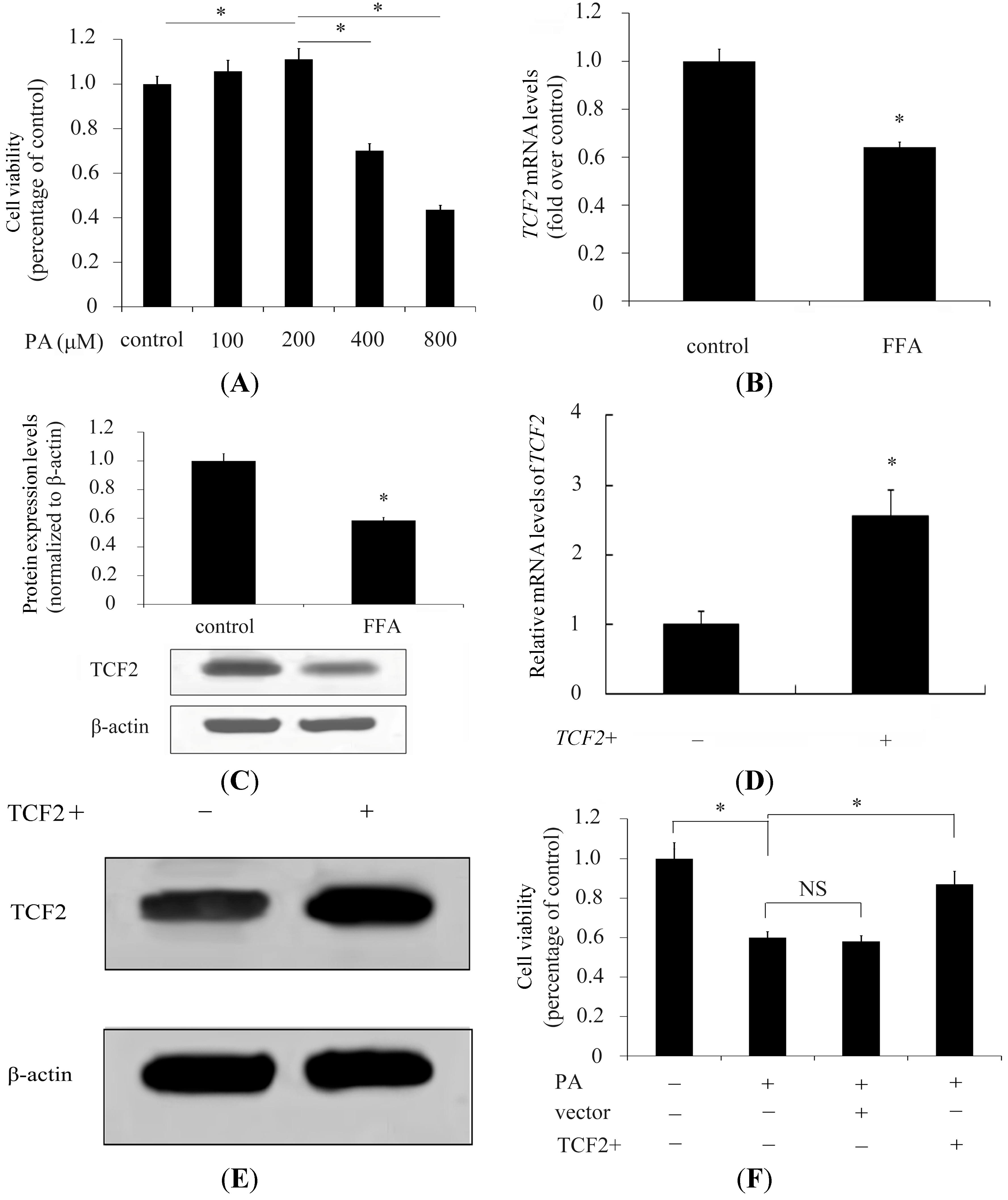
2.1. Transcription Factor 2 (TCF2) Relieved Free Fatty Acids (FFA) Induced Inhibitory Effect on INS-1 Cell Viability
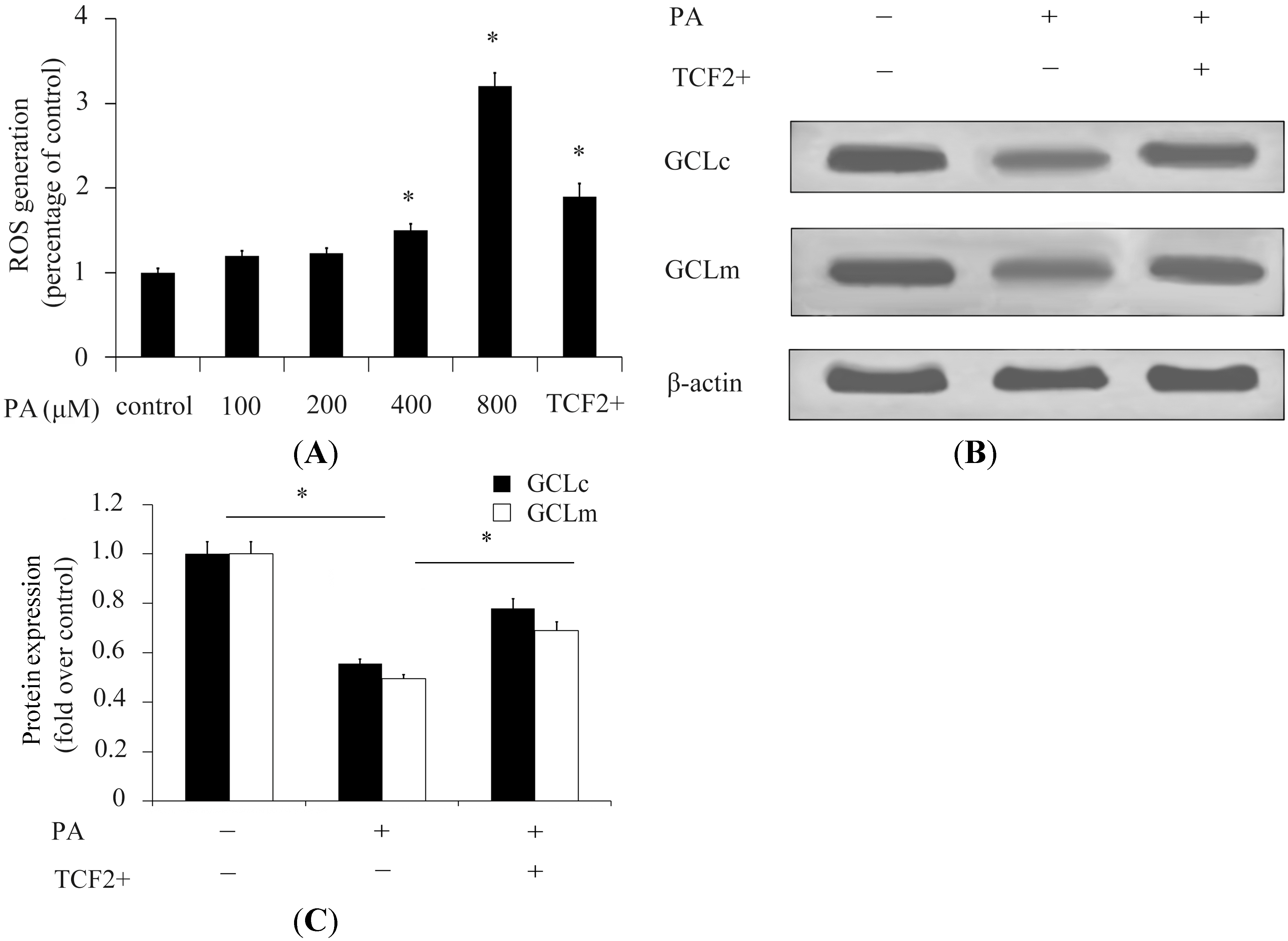
2.2. TCF2 Inhibited Reactive Oxygen Species (ROS) Production Stimulated by High Concentrations of Palmitic Acid (PA)


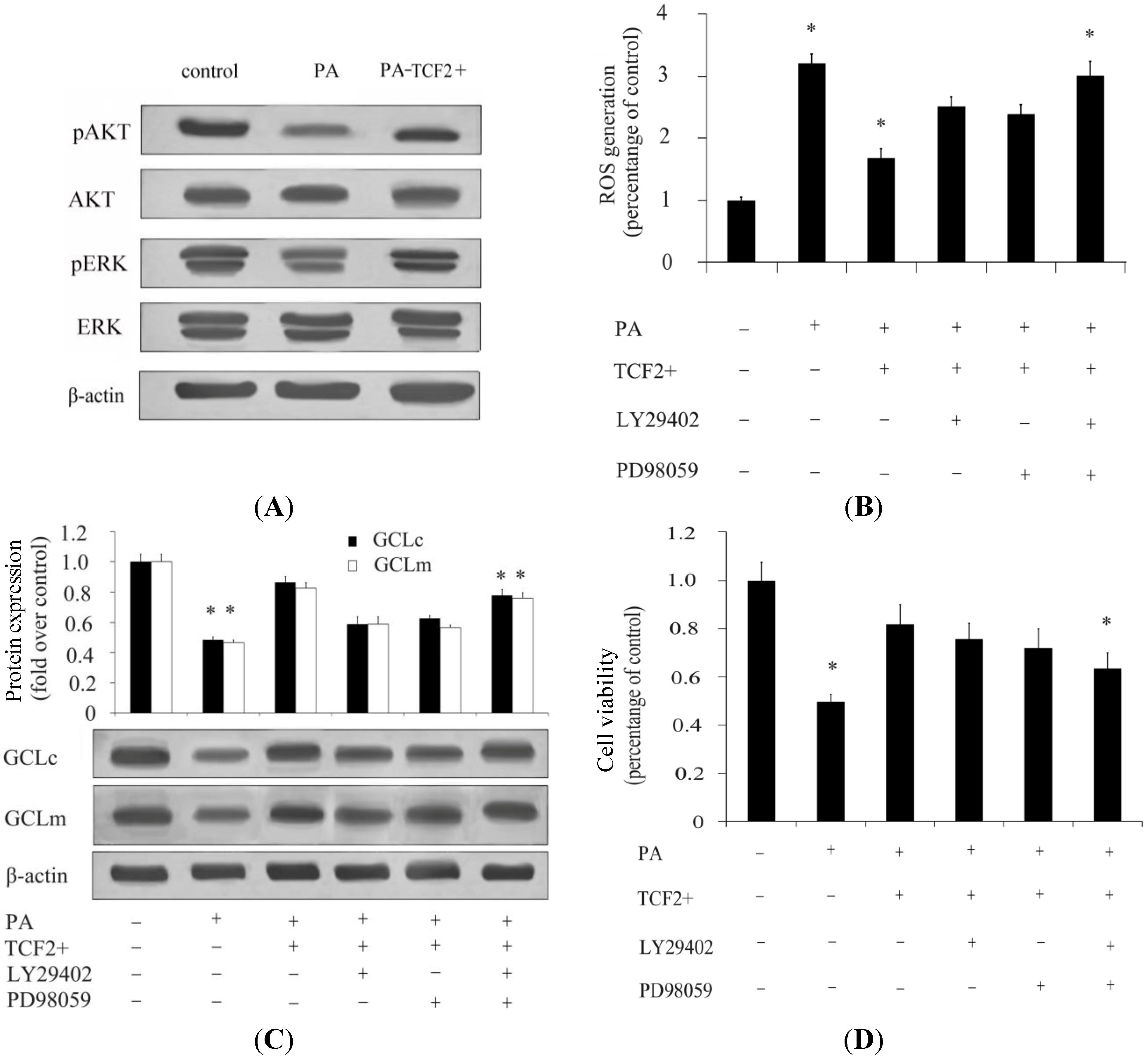
2.3. TCF2 Positively Regulated PI3K/AKT and MAPK/ERK Signaling Pathways

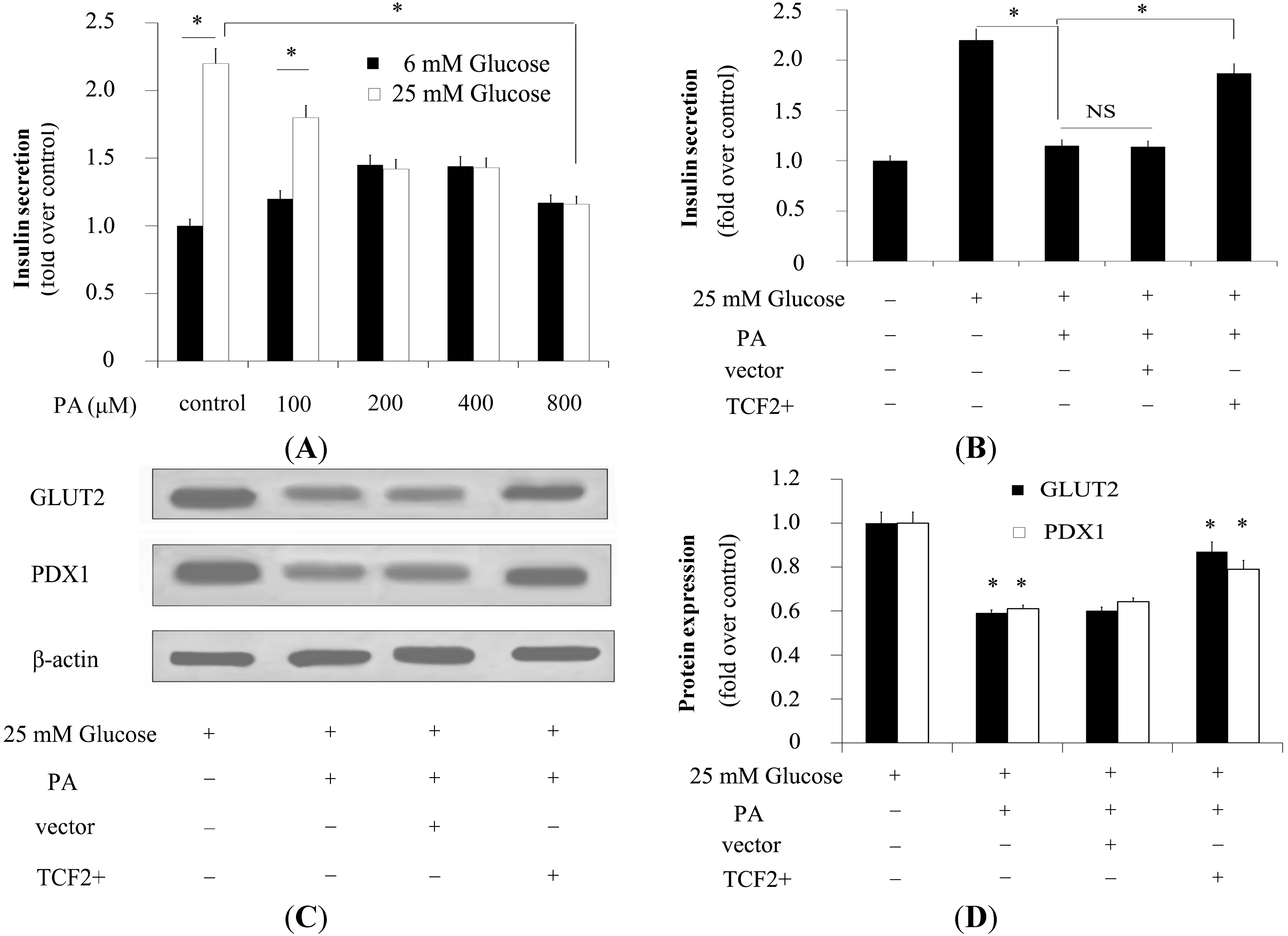
2.4. TCF2 Relieved FFA-Abrogated Insulin Secretion
2.5. TCF2 Positively Modulated Expression of Insulin Secretion-Related Molecules
2.6. TCF2 Positively Regulated Insulin Secretion via JNK Signaling


3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability
4.3. ROS Measurement
4.4. Adenovirus Infection
4.5. Insulin Secretion Assay
4.6. Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR).
4.7. Western Blotting
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eckel, R.H.; Kahn, S.E.; Ferrannini, E.; Goldfine, A.B.; Nathan, D.M.; Schwartz, M.W.; Smith, R.J.; Smith, S.R. Obesity and type 2 diabetes: What can be unified and what needs to be individualized? Diabetes Care 2011, 96, 1654–1663. [Google Scholar]
- Grupe, A.; Hultgren, B.; Ryan, A.; Ma, Y.H.; Bauer, M.; Stewart, T.A. Transgenic knockouts reveal a critical requirement for pancreatic β-cell glucokinase in maintaining glucose homeostasis. Cell 1995, 83, 69–78. [Google Scholar] [CrossRef]
- Ozasa, R.; Okada, T.; Nadanaka, S.; Nagamine, T.; Zyryanova, A.; Harding, H.; Ron, D.; Mori, K. The antipsychotic olanzapine induces apoptosis in insulin-secreting pancreatic β cells by blocking PERK-mediated translational attenuation. CellStruct. Funct. 2013, 8, 72–85. [Google Scholar]
- Buchanan, T.A. Pancreatic β-cell loss and preservation in type 2 diabetes. Clin. Ther. 2003, 25, 32–46. [Google Scholar] [CrossRef]
- Pipeleers, D.; Ling, Z. Pancreatic β-cells in insulin-dependent diabetes. Diabetes Metab. Rev. 1992, 8, 209–227. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Boden, G.; Shulman, G. Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and β-cell dysfunction. Eur. J. Clin. Investig. 2002, 32, 14–23. [Google Scholar] [CrossRef]
- Ayvaz, G.; Törüner, F.B.; Karakoc, A.; Yetkin, I.; Fakir, N.; Arslan, M. Acute and chronic effects of different concentrations of free fatty acids on the insulin secreting function of islets. Diabetes Metab. 2002, 28, 3S7–3S12. [Google Scholar]
- Borel, A.-L.; Boulet, G.; Nazare, J.-A.; Smith, J.; Alméras, N.; Tremblay, A.; Bergeron, J.; Poirier, P.; Carpentier, A.C.; Després, J.-P. Improved plasma FFA/insulin homeostasis is independently associated with improved glucose tolerance after a one-year lifestyle intervention in viscerally obese men. Diabetes Care 2013, 44, 28–37. [Google Scholar]
- Pedersen, K.B.; Chhabra, K.H.; Nguyen, V.K.; Xia, H.; Lazartigues, E. The transcription factor HNF1α induces expression of angiotensin-converting enzyme 2 (ACE2) in pancreatic islets from evolutionarily conserved promoter motifs. BBA Gene Regul. Mech. 2013, 1829, 1225–1235. [Google Scholar]
- Zhang, X.; Qiao, H.; Zhao, Y.; Wang, X.; Sun, H.M.; Liu, A.; Xu, L.; Sun, D.L.; Jin, Y.; Yu, Y.; et al. Association of single nucleotide polymorphisms in TCF2 with type 2 diabetes susceptibility in a Han Chinese population. PLoS One 2012, 7, 52938–52947. [Google Scholar]
- Tudorache, E.; Sellier-Leclerc, A.L.; Lenoir, M.; Toubiana, N.; Bensman, A.; Bellanne-Chantelot, C.; Ulinski, T. Childhood onset diabetes posttransplant in a girl with TCF2 mutation. Pediatr. Diabetes 2012, 13, 35–39. [Google Scholar] [CrossRef]
- Ancelle, D.; Hecart, A.C.; Gaillard, D.; Bertin, E.; Delemer, B. Deletions of TCF2 gene in Rokitansky syndrome (MRKH): A new candidate gene? About two new cases. Diabetes Care 2013, 12, 524–539. [Google Scholar]
- Haldorsen, I.S.; Vesterhus, M.; Raeder, H.; Jensen, D.K.; Søvik, O.; Molven, A.; Njølstad, P.R. Lack of pancreatic body and tail in HNF1B mutation carriers. Diabet. Med. 2008, 25, 782–787. [Google Scholar] [CrossRef]
- Johnstone, K.A.; Diakogiannaki, E.; Dhayal, S.; Morgan, N.G.; Harries, L.W. Dysregulation of Hnf1b gene expression in cultured β-cells in response to cytotoxic fatty acid. JOP J. Pancreas 2011, 12, 6–10. [Google Scholar]
- Teperino, R. Genetic Alterations in Type 2 Diabetes: Rregulation of PED/PEA-15 Gene Expression. Ph.D. Thesis, Universitá degli Studi di Napoli, Napoli, Italy, 2008. [Google Scholar]
- Baldwin, A.C.; Green, C.D.; Olson, L.K.; Moxley, M.A.; Corbett, J.A. A role for aberrant protein palmitoylation in FFA-induced ER stress and β-cell death. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1390–E1398. [Google Scholar] [CrossRef]
- Yuan, H.P.; Zhang, X.Y.; Huang, X.Q.; Lu, Y.G.; Tang, W.Q.; Man, Y.; Wang, S.; Xi, J.Z.; Li, J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of β-cells via JNK, p38 MAPK and p53 pathways. PLoS One 2010, 5, e15726. [Google Scholar]
- Tarantino, G.; Caputi, A. JNKs, insulin resistance and inflammation: A possible link between NAFLD and coronary artery disease. World J. Gastroenterol. 2011, 17, 3785–3794. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün1, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Gagnon, C.; Lu, Z.X.; Magliano, D.J.; Dunstan, D.W.; Shaw, J.E.; Zimmet, P.Z.; Sikaris, K.; Grantham, N.; Ebeling, P.R.; Daly, R.M. Serum 25-hydroxyvitamin D, calcium Intake, and risk of type 2 diabetes after 5 years results from a national, population-based prospective study (the Australian Diabetes, Obesity and Lifestyle Study). Diabetes Care 2011, 34, 1133–1138. [Google Scholar] [CrossRef]
- Kharroubi, I.; Ladriere, L.; Cardozo, A.K.; Dogusan, Z.; Cnop, M.; Eizirik, D.L. Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms: Role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology 2004, 145, 5087–5096. [Google Scholar] [CrossRef]
- Subauste, A.R.; Burant, C.F. Role of FoxO1 in FFA-induced oxidative stress in adipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 159–164. [Google Scholar] [CrossRef]
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.O.; Procopio, J.; Morgan, D.; Oliveira-Emilio, H.C.; Carpinelli, A.R.; Curi, R. Diabetes associated cell stress and dysfunction: Role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 2007, 583, 9–24. [Google Scholar] [CrossRef]
- Grønning, L.M.; Cederberg, A.; Miura, N.; Enerbäck, S.; Taskén, K. Insulin and TNFα induce expression of the forkhead transcription factor gene Foxc2 in 3T3-L1 adipocytes via PI3K and ERK 1/2-dependent pathways. Mol. Endocrinol. 2002, 16, 873–883. [Google Scholar]
- Chen, Y.W.; Huang, C.F.; Tsai, K.S.; Yang, R.S.; Yen, C.C.; Yang, C.Y.; Lin-Shiau, S.Y.; Liu, S.H. The role of phosphoinositide 3-kinase/Akt signaling in low-dose mercury-induced mouse pancreatic β-cell dysfunction in vitro and in vivo. Diabetes 2006, 55, 1614–1624. [Google Scholar] [CrossRef]
- Youl, E.; Bardy, G.; Magous, R.; Cros., G.; Sejalon, F.; Virsolvy, A.; Richard, S.; Quignard, J.F.; Gross, R.; Petit, P.; et al. Qercetin potentiates insulin secretion and protects INS-1 pancreatic β-cells against oxidative damage via the ERK1/2 pathway. Br. J. Pharmacol. 2010, 161, 799–814. [Google Scholar] [CrossRef]
- Zhang, Y.; Moerkens, M.; Ramaiahgari, S.; de Bont, H.; Price, L.; Meerman, J.; van de Water, B. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 2011, 13, 52–65. [Google Scholar] [CrossRef]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Investig. 2011, 121, 2118–2125. [Google Scholar] [CrossRef]
- Feng, Z.-C.; Riopel, M.; Li, J.; Donnelly, L.; Wang, R. Downregulation of Fas activity rescues early onset of diabetes in c-KitWv/+ mice. Am. J. Physiol. Endocrinol. Metab. 2013, 304, 557–565. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Chen, M.Z.; Olefsky, J.M.; Marth, J.D. Pathway to diabetes through attenuation of pancreatic β-cell glycosylation and glucose transport. Nat. Med. 2011, 17, 1067–1075. [Google Scholar] [CrossRef]
- Guillam, M.-T.; Hümmler, E.; Schaerer, E.; Wu, J.-Y.; Birnbaum, M.J.; Beermann, F.; Schmidt, A.; Dériaz, N.; Thorens, B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef]
- Holland, A.M.; Garcia, S.; Naselli, G.; MacDonald, R.J.; Harrison, L.C. The Parahox gene Pdx1 is required to maintain positional identity in the adult foregut. Int. J. Dev. Biol. 2012, 57, 391–398. [Google Scholar]
- Guo, L.L.; Inada, A.I.; Aguayo-Mazzucato, C.; Hollister-Lock, J.; Fujitani, Y.; Weir, G.C.; Wright, C.V.E.; Sharma, A.; Bonner-Weir, S. PDX1 in ducts is not required for postnatal formation of β-cells but is necessary for their subsequent maturation. Diabetes 2013, 62, 3459–3468. [Google Scholar] [CrossRef]
- Biteau, B.; Karpac, J.; Hwangbo, D.; Jasper, H. Regulation of Drosophila lifespan by JNK signaling. Exp. Gerontol. 2011, 46, 349–354. [Google Scholar] [CrossRef]
- De Souza, L.F.; Barreto, F.; da Silva, E.G.; Andrades, M.E.; Guimarães, E.L.M.; Behr, G.A.; Moreira, J.C.F.; Bernard, E.A. Regulation of LPS stimulated ROS production in peritoneal macrophages from alloxan-induced diabetic rats: Involvement of high glucose and PPARγ. Life Sci. 2007, 81, 153–159. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Quan, X.; Zhang, L.; Li, Y.; Liang, C. TCF2 Attenuates FFA-Induced Damage in Islet β-Cells by Regulating Production of Insulin and ROS. Int. J. Mol. Sci. 2014, 15, 13317-13332. https://doi.org/10.3390/ijms150813317
Quan X, Zhang L, Li Y, Liang C. TCF2 Attenuates FFA-Induced Damage in Islet β-Cells by Regulating Production of Insulin and ROS. International Journal of Molecular Sciences. 2014; 15(8):13317-13332. https://doi.org/10.3390/ijms150813317
Chicago/Turabian StyleQuan, Xiaojuan, Lin Zhang, Yingna Li, and Chunlian Liang. 2014. "TCF2 Attenuates FFA-Induced Damage in Islet β-Cells by Regulating Production of Insulin and ROS" International Journal of Molecular Sciences 15, no. 8: 13317-13332. https://doi.org/10.3390/ijms150813317