Identification of Bacterial Community Composition in Freshwater Aquaculture System Farming of Litopenaeus vannamei Reveals Distinct Temperature-Driven Patterns

Abstract
:1. Introduction
2. Results and Discussion
2.1. DGGE-Based Bacterial Population Profiles in the FASFL


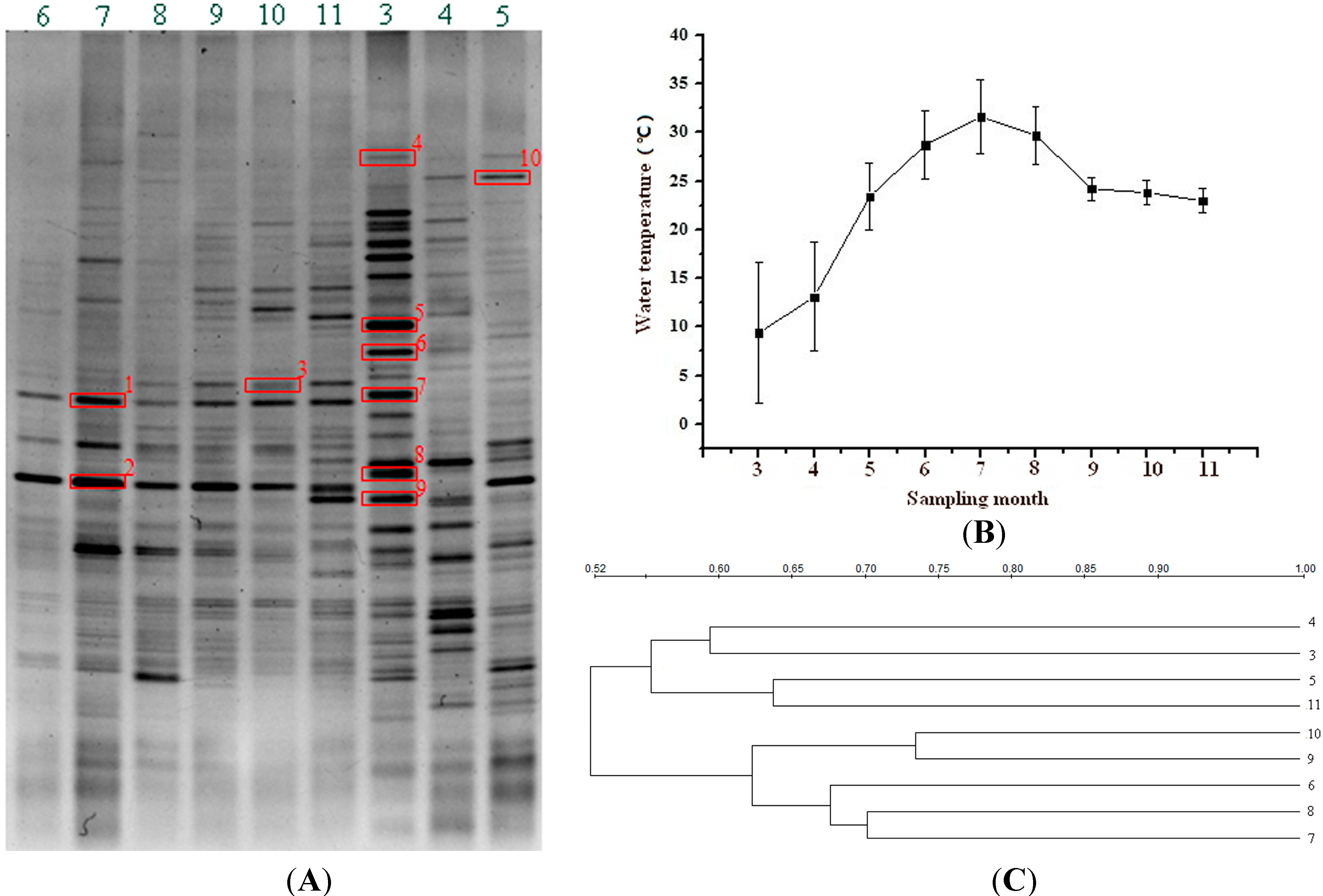{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Band | Clustering Group | Length (bp) | Closest Relative and Database Accession Number | Identity (%) | Taxonomic Description | |
|---|---|---|---|---|---|---|
| B1 | III | 169 | Uncultured Rickettsiales bacterium clone FWB6C1-73, KF583165.1 | 100% | α-Proteobacteria | |
| B2 | III | 174 | Uncultured Micrococcineae bacterium clone D7N78, KC006224.1 | 100% | Actinobacteria | |
| B3 | III | 194 | Uncultured Methylophilus sp. clone JA127_2010-09-15, JN866934.1 | 100% | β-Proteobacteria | |
| 169 | Sphingomonas oligophenolica strain R2A-AUG-EA-11, JX237432.1, KC836618.1 | 100% | α-Proteobacteria | |||
| B4 | I | 194 | Pediococcus ethanolidurans strain RU12-4 | 100% | Firmicutes | |
| B5 | I | 189 | Uncultured Bacteroidetes bacterium, FR647662.1 | 100% | Bacteroidetes | |
| 189 | Uncultured Cryomorphaceae bacterium clone Jab PL2W2H12, HM486317.1 | 100% | Bacteroidetes | |||
| B6 | I | 174 | Actinobacterium MS-B-64, FJ460153.1 | 100% | Actinobacteria | |
| B7 | I | 189 | Uncultured Bacteroidetes bacterium clone XSLJ052, KC246401.1 | 100% | Bacteroidetes | |
| B8 | I | 174 | Uncultured actinobacterium clone FF1G3, EU117678.1 | 100% | Actinobacteria | |
| 174 | Uncultured actinobacterium clone B12-88, JN371245.1 | 100% | Actinobacteria | |||
| 174 | Actinobacterium SCGC AAA043-A09, HQ663377.1 | 100% | Actinobacteria | |||
| B9 | I | 174 | Uncultured Micrococcineae bacterium clone D7N78, KC006224.1 | 100% | Actinobacteria | |
| B10 | II | 174 | Uncultured Intrasporangiaceae bacterium clone M7N57, KC006381.1 | 100% | Actinobacteria | |
2.2. Environmental Temperature and Bacterial Population in the FASFL

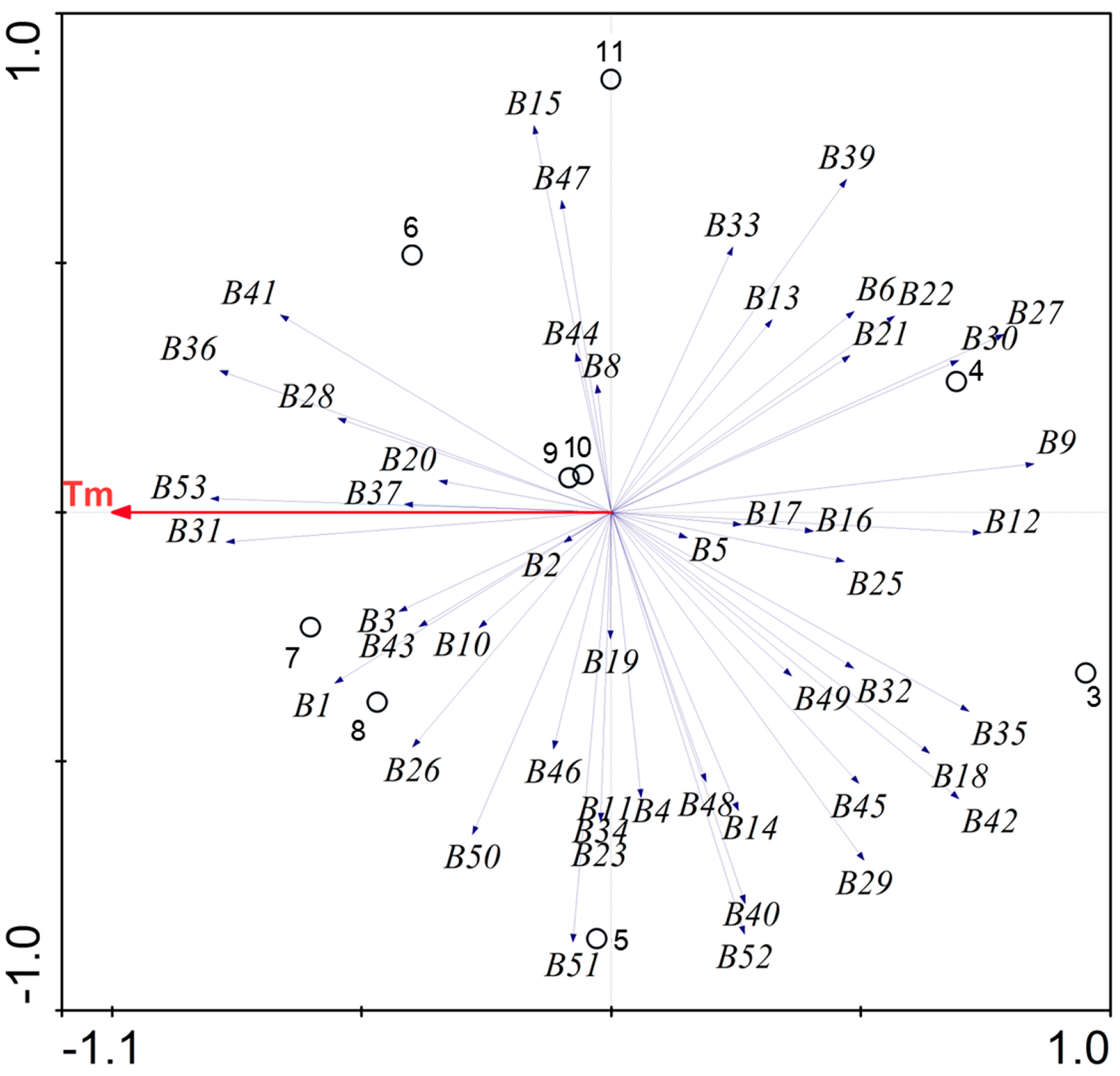
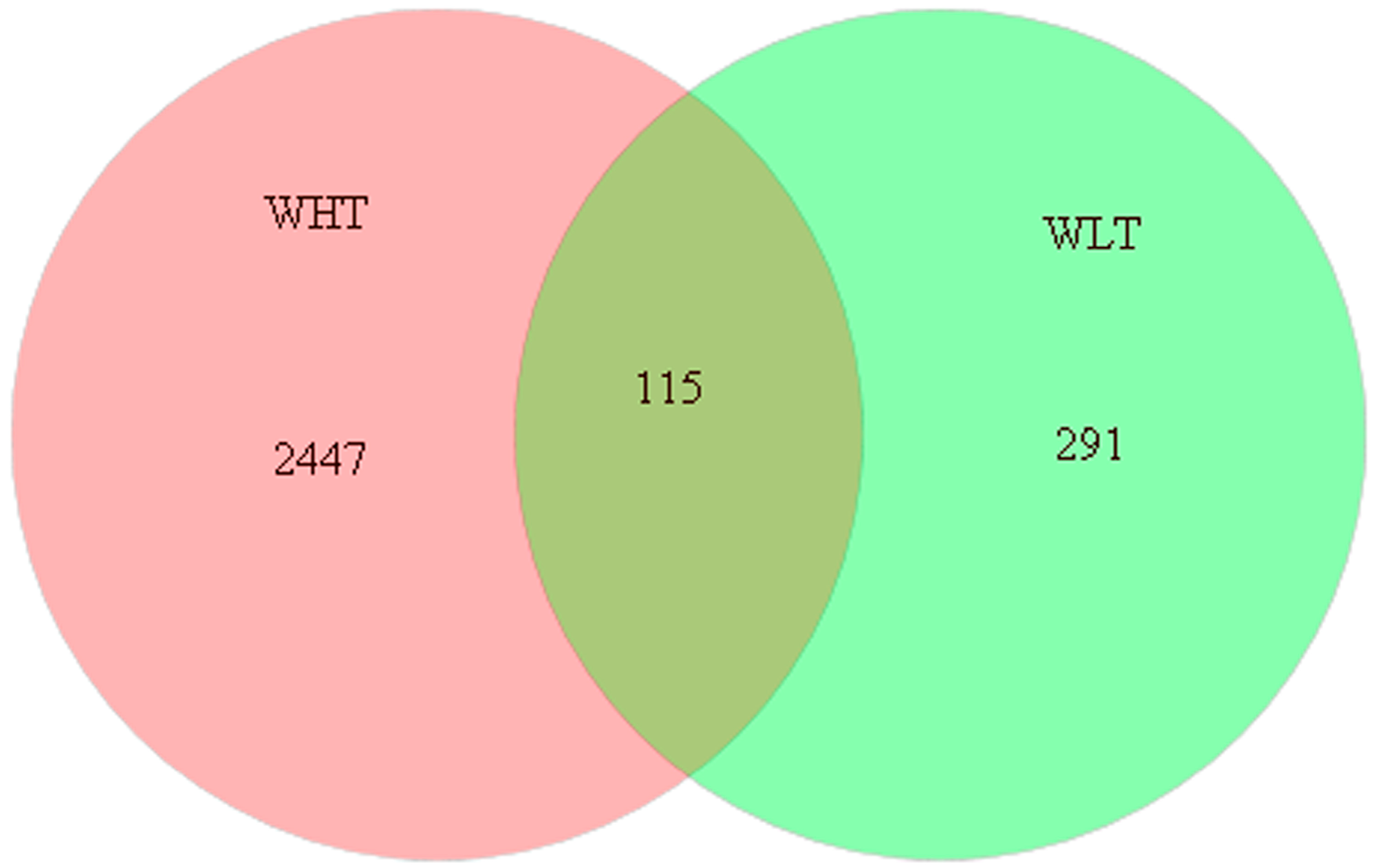
2.3. Overall Bacterial Community Composition in the FASFL
2.3.1. Bacterial Richness and Diversity
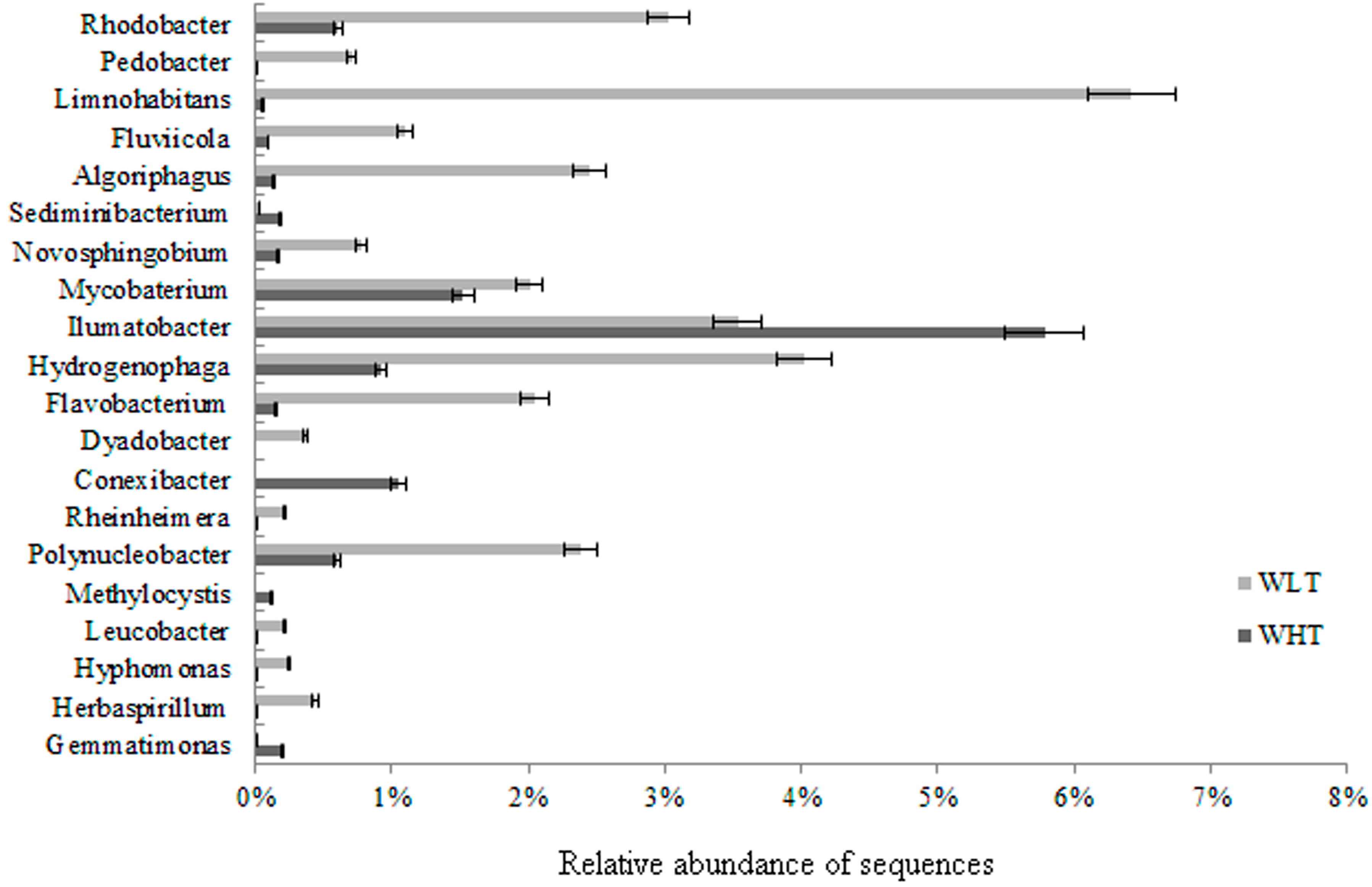
2.3.2. Bacterial Community Composition



2.3.3. Potential Pathogenic Bacteria
| Bacterial Pathogen a | Disease | Source | RAS (%) b | ||
|---|---|---|---|---|---|
| Environment | Main Hosts | WHT | WLT | ||
| Aeromonas hydrophila | Motile aeromonads septicaemia, cholangitis | Freshwater, brackish water, biosolid | Catfish, carp, trout, eel, sturgeon, tilapia, bass | 0.0109 | 0.0054 |
| Aeromonas caviae | Speticaemia, gastroenteritis, cholangitis | Freshwater, brackish water, soil, biosolid, agricultural products, | Fish, shrimp, frog, soft-shelled turtle | 0.0054 | 0 |
| Aeromonas veronii | Speticaemia, gastroenteritis, cholangitis | Freshwater, biosolid | Human, mosquitos, leeches | 0 | 0.0054 |
| Bacillus anthracis | Anthrax | Natural and processed water sources, sewage, biosolid | Human | 0.0054 | 0.0054 |
| Flavobacterium johnsoniae | False columnaris | Natural water sources, fish cultures | Barramundi | 0 | 0.0054 |
| Mycobacterium avium | Mycobacterium avium complex | Natural water sources, soil, biosolid | Human, farm animals, birds | 0.0054 | 0 |
| Mycobacterium fortuitum | Osteomyelitis | River, lake, tap water, soil, dust, biosolid | Human, cattle, frog, other animals | 0.0217 | 0.0163 |
| Mycobacterium marinum | Mycobacteriosis | Natural water sources, fish cultures | Atlantic salmo, Seabass, turbot | 0.0054 | 0 |
| Serratia marcescens | Conjunctivitis, keratitis, endophthalmitis, tear duct infections | Natural water sources, soil, biosolid | Human, plants, animals | 0 | 0.0109 |
| Pseudomonas anguilliseptica | Pseudomonadiasis, Winter disease | Natural water sources, fish cultures | Human, seabream, eel, turbot, ayu | 0.0163 | 0.0109 |
| Vibrio cholerae | Vibriosis | Natural water sources, fish cultures, biosolid | Human, croaker fish, puffer fish, grouper, cod, shrimp, big-scale sand smelt, flounder, abalone, seabream, salmon, sweetfish, sheatfish, catfish | 0 | 0.0054 |
3. Experimental Section
3.1. Sample Collection and Bacterial Genomic DNA Extraction
3.2. PCR-DGGE and Data Analysis
3.3. Sequencing and Phylogenetic Analysis
3.4. Pyrosequencing and Data Analysis
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations. FAO Yearbook 2010: Fishery and Aquaculture Statistics; FAO: Rome, Italy, 2012. [Google Scholar]
- Liao, C.; Chien, Y.-H. The Pacific white shrimp, Litopenaeus vannamei, in Asia: The world’s most widely cultured alien crustacean. In The Wrong Place-Alien Marine Crustaceans: Distribution, Biology and Impacts; Galil, B.S., Clark, P.F., Carlton, J.T., Eds.; Invading Nature-Springer Series in Invasion Ecology: Dordrecht, The Netherlands, 2011; Volume 6, pp. 489–519. [Google Scholar]
- Food and Agriculture Organization of the United Nations. FAO Yearbook 2009: Fishery and Aquaculture Statistics. FAO: Rome, Italy, 2011. [Google Scholar]
- Thompson, F.L.; Iida, T.; Swings, J. Biodiversity of Vibrios. Microbiol. Mol. Biol. R. 2004, 68, 403–431. [Google Scholar] [CrossRef]
- Lavilla-Pitogo, C.R.; Leano, E.M.; Paner, M.G. Mortalities of pond-cultured juvenile shrimp, Penaeus monodon, associated with dominance of luminescent Vibrios in the rearing environment. Aquaculture 1998, 164, 337–349. [Google Scholar] [CrossRef]
- Sung, H.-H.; Hsu, S.-F.; Chen, C.-K.; Ting, Y.-Y.; Chao, W.-L. Relationships between disease outbreak in cultured tiger shrimp (Penaeus monodon) and the composition of Vibrio communities in pond water and shrimp hepatopancreas during cultivation. Aquaculture 2001, 192, 101–110. [Google Scholar] [CrossRef]
- Israngkura, A.; Sae-Hae, S. A review of the economic impacts of aquatic animal disease. In Primary Aquatic Animal Health Care in Rural, Small-Scale, Aquaculture Development; Arthur, J.R., Phillips, M.J., Subasinghe, R.P., Reantaso, M.B., MacRae, I.H., Eds.; FAO Fish. Tech.: Rome, Italy, 2002; Volume 406, pp. 253–286. [Google Scholar]
- Bourne, D.G.; Young, N.; Nicole Webster, N.; Payne, M.; Salmon, M.; Demel, S.; Hall, M. Microbial community dynamics in a larval aquaculture system of the tropical rock lobster, Panulirus ornatus. Aquaculture 2004, 242, 31–51. [Google Scholar] [CrossRef]
- Zhou, J.; Fang, W.; Yang, X.; Zhou, S.; Hu, L.; Li, X.; Qi, X.; Su, H.; Xie, L.A. Nonluminescent and highly virulent Vibrio harveyi strain is associated with “Bacterial White Tail Disease” of Litopenaeus vannamei shrimp. PLoS One 2012, 7, e29961. [Google Scholar]
- Wilcox, B.A.; Colwell, R.R. Emerging and reemerging infectious diseases: Biocomplexity as an interdisciplinary paradigm. EcoHealth 2005, 2, 244–257. [Google Scholar] [CrossRef]
- Hasson, K.W.; Wyld, E.M.; Fan, Y.; Lingsweiller, S.W.; Wiener, S.J.; Cheng, J.; Varner, P.W. Streptococcosis in farmed Litopenaeus vannamei: A new emerging bacterial disease of penaeid shrimp. Dis. Aquat. Organ. 2009, 86, 93–106. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies-the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Payne, M.S.; Hall, M.R.; Sly, L.; Bourne, D.G. Microbial diversity within early-stage cultured Panulirus ornatus phyllosomas. Appl. Environ. Microbiol. 2007, 73, 1940–1951. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.N.; Barnes, S.; Ogle, J.; Grimes, D.J. Microbial community analysis of water, foregut, and hindgut during growth of Pacific white shrimp, Litopenaeus vannamei, in closed-system aquaculture. J. World Aquacult. Soc. 2008, 39, 251–258. [Google Scholar] [CrossRef]
- Sakami, T.; Fujioka, Y.; Shimoda., T. Comparison of microbial community structures in intensive and extensive shrimp culture ponds and a mangrove area in Thailand. Fish. Sci. 2008, 74, 889–898. [Google Scholar] [CrossRef]
- Van der Meeren, T.; Brunvold, L.; Sandaa, R.-A.; Bergh, O.; Castberg, T.; Thyrhaug, R.; Mangor-Jensen, A. Water quality and microbial community structure in juvenile Atlantic cod (Gadus morhua L.) cultures. Aquaculture 2011, 316, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Fu, L.; Deng, B.; Liang, Q.; Zheng, J.; Sun, J.; Zhu, H.; Peng, L.; Wang, Y.; Shen, W.; et al. Bacillus subtilis SC02 supplementation causes alterations of the microbial diversity in grass carp water. World J. Microbiol. Biotechnol. 2013, 29, 1645–1653. [Google Scholar] [CrossRef]
- Zeng, Y.; Ma, Y.; Wei, C.; Jiao, N.; Tang, K.; Wu, Z.; Jian, J. Bacterial diversity in various coastal mariculture ponds in Southeast China and in diseased eels as revealed by culture and culture-independent molecular techniques. Aquacult. Res. 2010, 41, e172–e186. [Google Scholar] [CrossRef]
- Youssef, N.H.; Couger, M.B.; Elshahed, M.S. Fine-scale bacterial beta diversity within a complex ecosystem (Zodletone Spring, OK, USA): The role of the rare biosphere. PLoS One 2010, 5, e12414. [Google Scholar] [CrossRef]
- Han, D.; Kang, I.; Ha, H.K.; Kim, H.C.; Kim, O.-S.; Lee, B.Y.; Cho, J.-C.; Hur, H.-G.; Lee, Y.K. Bacterial communities of surface mixed layer in the Pacific sector of the western Arctic ocean during sea-ice melting. PLoS One 2014, 9, e86887. [Google Scholar]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Nat. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef]
- Vaz-Moreira, I.; Egas, C.; Nunes, O.C.; Manaia, C.M. Culture-dependent and culture-independent diversity surveys target different bacteria: A case study in a freshwater sample. Anton. Leeuw. Int. J. G. 2011, 100, 245–257. [Google Scholar] [CrossRef]
- Miller, S.R.; Strong, A.L.; Jones, K.L.; Ungerer, M.C. Bar-coded pyrosequencing reveals shared bacterial community properties along the temperature gradients of two alkaline hot springs in yellowstone national park. Appl. Environ. Microbiol. 2009, 75, 4565–4572. [Google Scholar] [CrossRef]
- Revetta, R.P.; Pemberton, A.; Lamendella, R.; Iker, B.; Santo Domingo, J.W. Identification of bacterial populations in drinking water using 16S rRNA-based sequence analyses. Water Res. 2010, 44, 1353–1360. [Google Scholar]
- Shanks, O.C.; Newton, R.J.; Kelty, C.A.; Huse, S.M.; Sogin, M.L.; McLellan, S.L. Comparison of the microbial community structures of untreated wastewaters from different geographic locales. Appl. Environ. Microbiol. 2013, 79, 2906–2913. [Google Scholar] [CrossRef]
- Beleneva, I.A.; Zhukova, N.V. Seasonal dynamics of cell numbers and biodiversity of marine heterotrophic bacteria inhabiting invertebrates and water ecosystems of the Peter the Great Bay, Sea of Japan. Microbiology 2009, 78, 369–375. [Google Scholar] [CrossRef]
- Zhang, M.; Yu, N.; Chen, L.; Jiang, C.; Tao, Y.; Zhang, T.; Chen, J.; Xue, D. Structure and seasonal dynamics of bacterial communities in three urban rivers in China. Aquat. Sci. 2012, 74, 113–120. [Google Scholar] [CrossRef]
- Rösel, S.; Allgaier, M.; Grossart, H.-P. Long-term characterization of free-living and particle-associated bacterial communities in lake Tiefwaren reveals distinct seasonal patterns. Microb. Ecol. 2012, 64, 571–583. [Google Scholar] [CrossRef]
- Jing, H.; Xia, X.; Suzuki, K.; Liu, H. Vertical profiles of bacteria in the tropical and subarctic oceans revealed by pyrosequencing. PLoS One 2013, 8, e79423. [Google Scholar] [CrossRef]
- Bakke, I.; Skjermo, J.; Vo, T.A.; Vadstein, O. Live feed is not a major determinant of the microbiota associated with cod larvae (Gadus morhua). Environ. Microbiol. Rep. 2013, 5, 537–548. [Google Scholar] [CrossRef]
- Nacke, H.; Thürme, A.; Wollherr, A.; Will, C.; Hodac, L.; Herold, N.; Schöning, I.; Schrumpf, M.; Daniel, R. Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS One 2011, 6, e17000. [Google Scholar] [CrossRef]
- Martins, P.; Cleary, D.F.R.; Pires, A.C.C.; Rodrigues, A.M.; Quintino, V.; Calado, R.; Gomes, N.C.M. Molecular analysis of bacterial communities and detection of potential pathogens in a recirculating aquaculture system for Scophthalmus maximus and Solea senegalensis. PLoS One 2013, 8, e80847. [Google Scholar]
- Dworkin, M.; Falkow, S.; Rosenberg, E.; Schleifer, K.H.; Stackebrandt, E. (Eds.) The Prokaryotes: A Handbook on the Biology of Bacteria; Springer-Verlag: New York, NY, USA, 2006; pp. 3–37.
- Matsumoto, A.; Kasai, H.; Matsuo, Y.; Ōmura, S.; Shizuri, Y.; Takahashi, Y. Ilumatobacter fluminis gen. nov., sp. nov., a novel actinobacterium isolated from the sediment of an estuary. J. Gen. Appl. Microbiol 2009, 55, 201–205. [Google Scholar] [CrossRef]
- Bibby, K.; Viau, E.; Peccia, J. Pyrosequencing of the 16S rRNA gene to reveal bacterial pathogen diversity in biosolids. Water Res. 2010, 44, 4252–4260. [Google Scholar] [CrossRef]
- Toranzo, A.E.; Magariños, B.; Romalde, J.L. A review of the main bacterial fish diseases in mariculture systems. Aquaculture 2005, 246, 37–61. [Google Scholar] [CrossRef]
- Pridgeon, J.W.; Klesius, P.H. Major bacterial diseases in aquaculture and their vaccine development. CAB Rev. 2012, 7, 048. [Google Scholar]
- Muyer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S Rrna. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar]
- Zhang, T.; Pan, Y.; Li, B.; Ou, J.; Zhang, J.; Chen, Y.; Peng, X.; Chen, L. Molecular cloning and antimicrobial activity of enterolysin A and helveticin J of bacteriolysins from metagenome of Chinese traditional fermented foods. Food Control 2013, 31, 499–507. [Google Scholar] [CrossRef]
- Clarke, K.R.; Green, R.H. Statistical design and analysis for a “biological effects” study. Mar. Ecol. Prog. Ser. 1988, 46, 213–226. [Google Scholar] [CrossRef]
- Ter Braak, C.J.F.; Šmilauer, P. CANOCO reference manual and CanoDraw for Windows user’s guide: Software for canonical community ordination (version 4.5). In Section on Permutation Methods. Microcomputer Power; Ithaca: New York, NY, USA, 2002. [Google Scholar]
- Schabereiter-Gurtner, C.; Maca, S.; Rölleke, S.; Nigl, K.; Lukas, J.; Hirschl, A.; Lubitz, W.; Barisani-Asenbauer, T. 16S rDNA-based identification of bacteria from conjunctival swabs by PCR and DGGE fingerprinting. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1164–1171. [Google Scholar]
- Shi, Y.; Pan, Y.; Li, B.; He, W.; She, Q.; Chen, L. Molecular cloning of a novel bioH gene from an environmental metagenome encoding a carboxylesterase with exceptional tolerance to organic solvents. BMC Biotechnol. 2013, 13, 13. [Google Scholar] [CrossRef]
- Song, Y.; Yu, P.; Li, B.; Pan, Y.; Zhang, X.; Cong, J.; Zhao, Y.; Wang, H.; Chen, L. The mosaic accessory gene structures of the SXT/R391-like integrative and conjugative elements derived from Vibrio spp. isolated from aquatic products and environment in the Yangtze River estuary, China. BMC Microbiol 2013, 13, 214. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. ntroducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Maidak, B.L.; Olsen, G.J.; Larsen, N.; Overbeek, R.; McCaughey, M.J.; Woese, C.R. The Ribosomal Database Project (RDP). Nucleic Acids Res. 1996, 24, 82–85. [Google Scholar] [CrossRef]
- Chao, A.; Bung, J. Estimating the number of species in a stochastic abundance model. Biometrics 2002, 58, 531–539. [Google Scholar] [CrossRef]
- Shannon, C.E.; Weaver, W. The mathematical theory of information. AT&T Tech. J 1949, 27, 379–423. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tang, Y.; Tao, P.; Tan, J.; Mu, H.; Peng, L.; Yang, D.; Tong, S.; Chen, L. Identification of Bacterial Community Composition in Freshwater Aquaculture System Farming of Litopenaeus vannamei Reveals Distinct Temperature-Driven Patterns. Int. J. Mol. Sci. 2014, 15, 13663-13680. https://doi.org/10.3390/ijms150813663
Tang Y, Tao P, Tan J, Mu H, Peng L, Yang D, Tong S, Chen L. Identification of Bacterial Community Composition in Freshwater Aquaculture System Farming of Litopenaeus vannamei Reveals Distinct Temperature-Driven Patterns. International Journal of Molecular Sciences. 2014; 15(8):13663-13680. https://doi.org/10.3390/ijms150813663
Chicago/Turabian StyleTang, Yuyi, Peiying Tao, Jianguo Tan, Haizhen Mu, Li Peng, Dandan Yang, Shilu Tong, and Lanming Chen. 2014. "Identification of Bacterial Community Composition in Freshwater Aquaculture System Farming of Litopenaeus vannamei Reveals Distinct Temperature-Driven Patterns" International Journal of Molecular Sciences 15, no. 8: 13663-13680. https://doi.org/10.3390/ijms150813663