Matrilin-3 Chondrodysplasia Mutations Cause Attenuated Chondrogenesis, Premature Hypertrophy and Aberrant Response to TGF-β in Chondroprogenitor Cells

 ,
, 


Abstract
:1. Introduction
2. Results
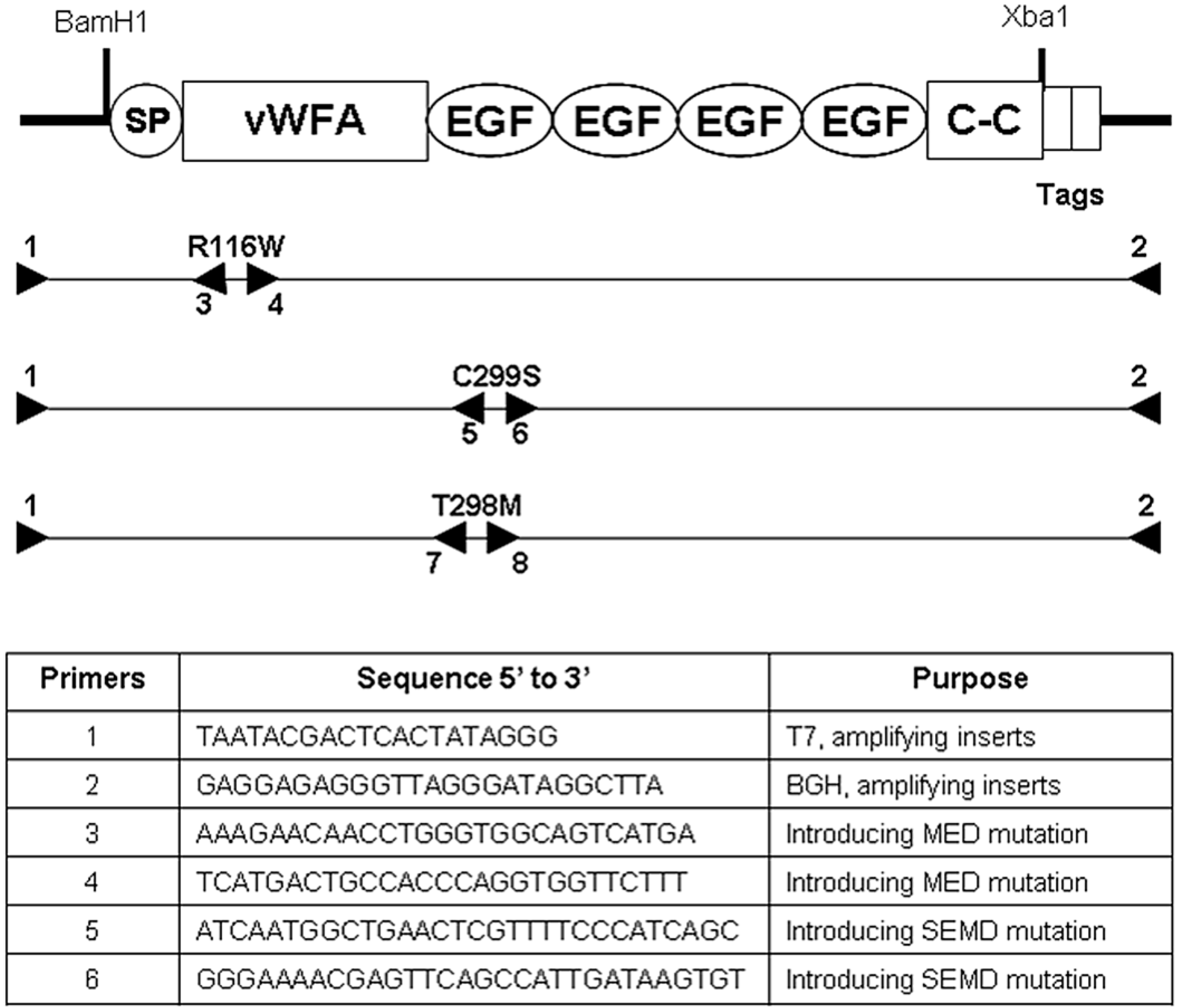
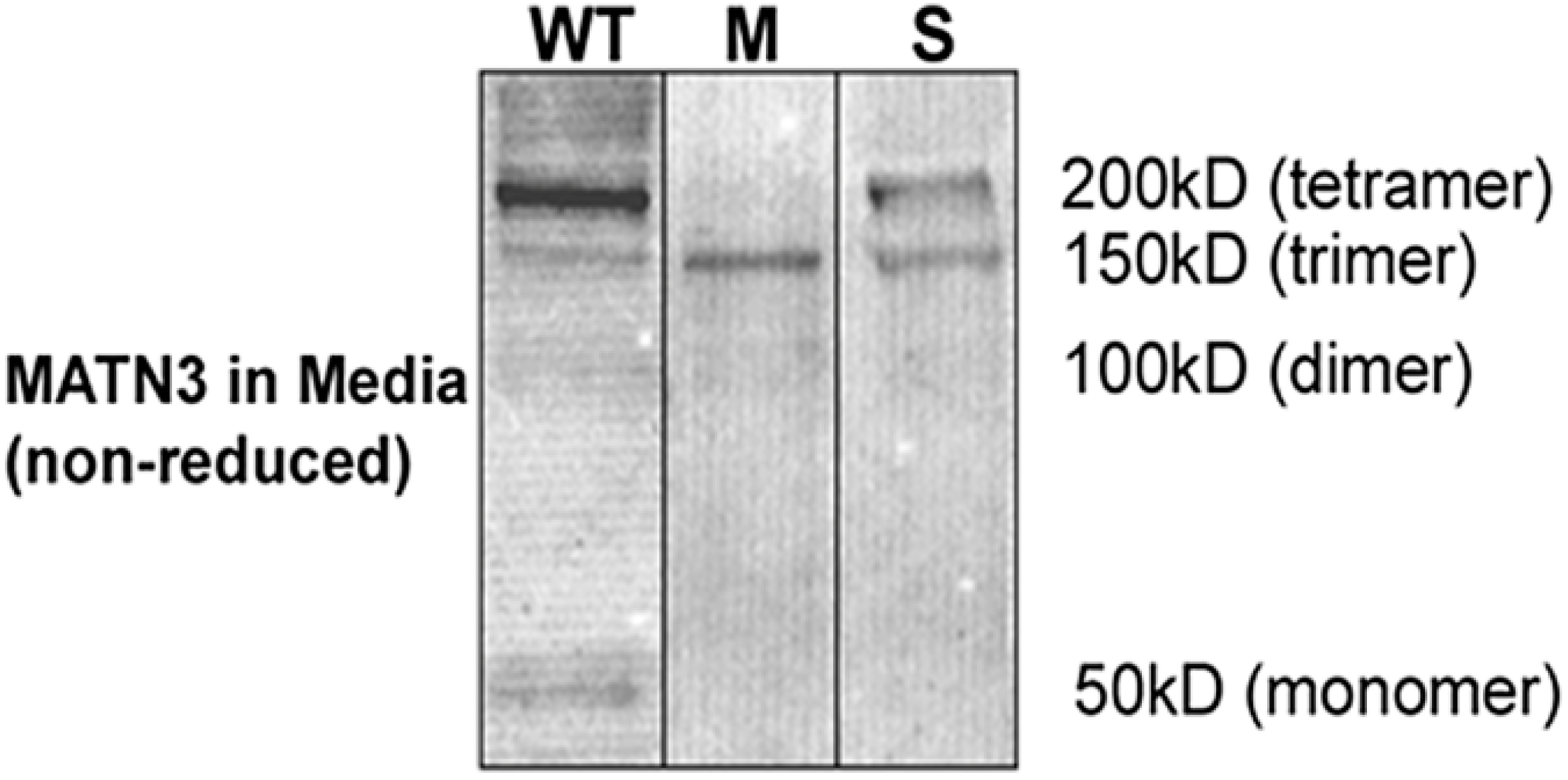
2.1. Establishment of Stable ATDC5 Cell Lines Harboring MED and SEMD Associated MATN3 Mutations


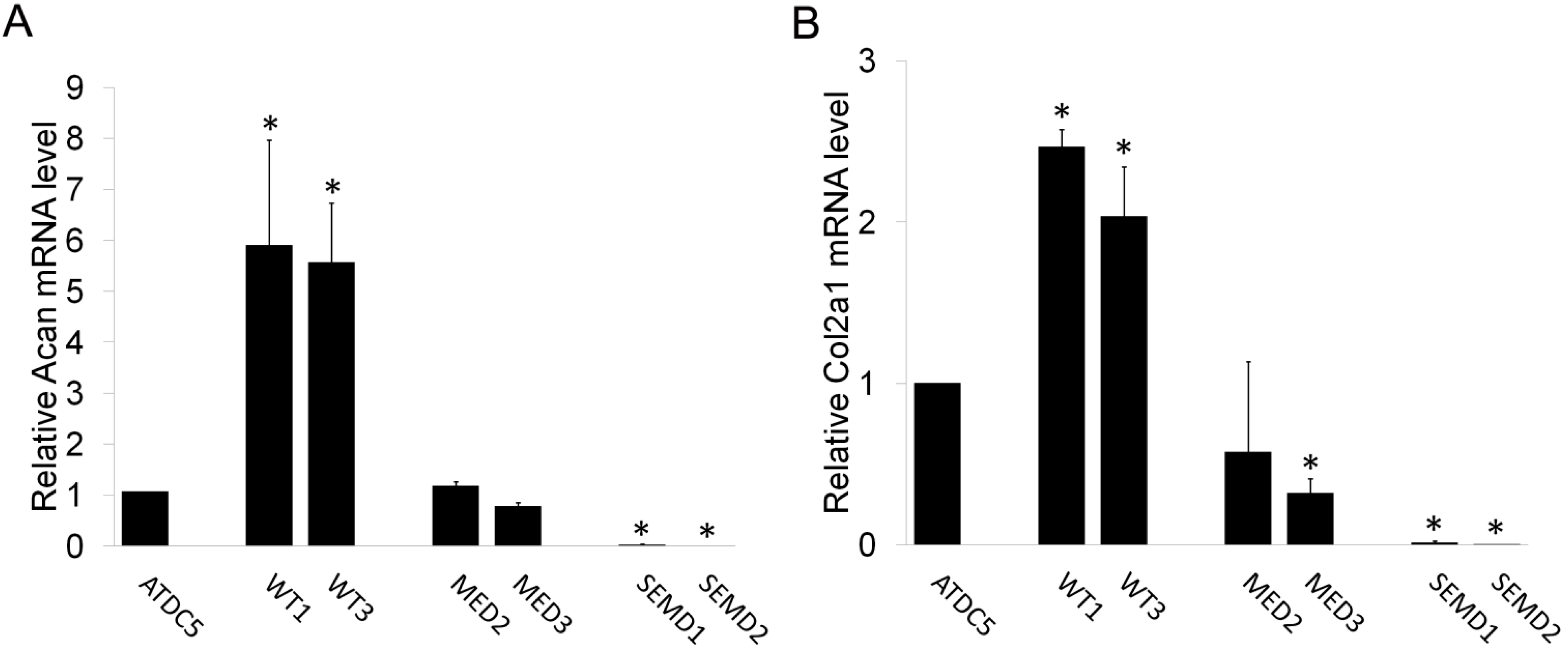
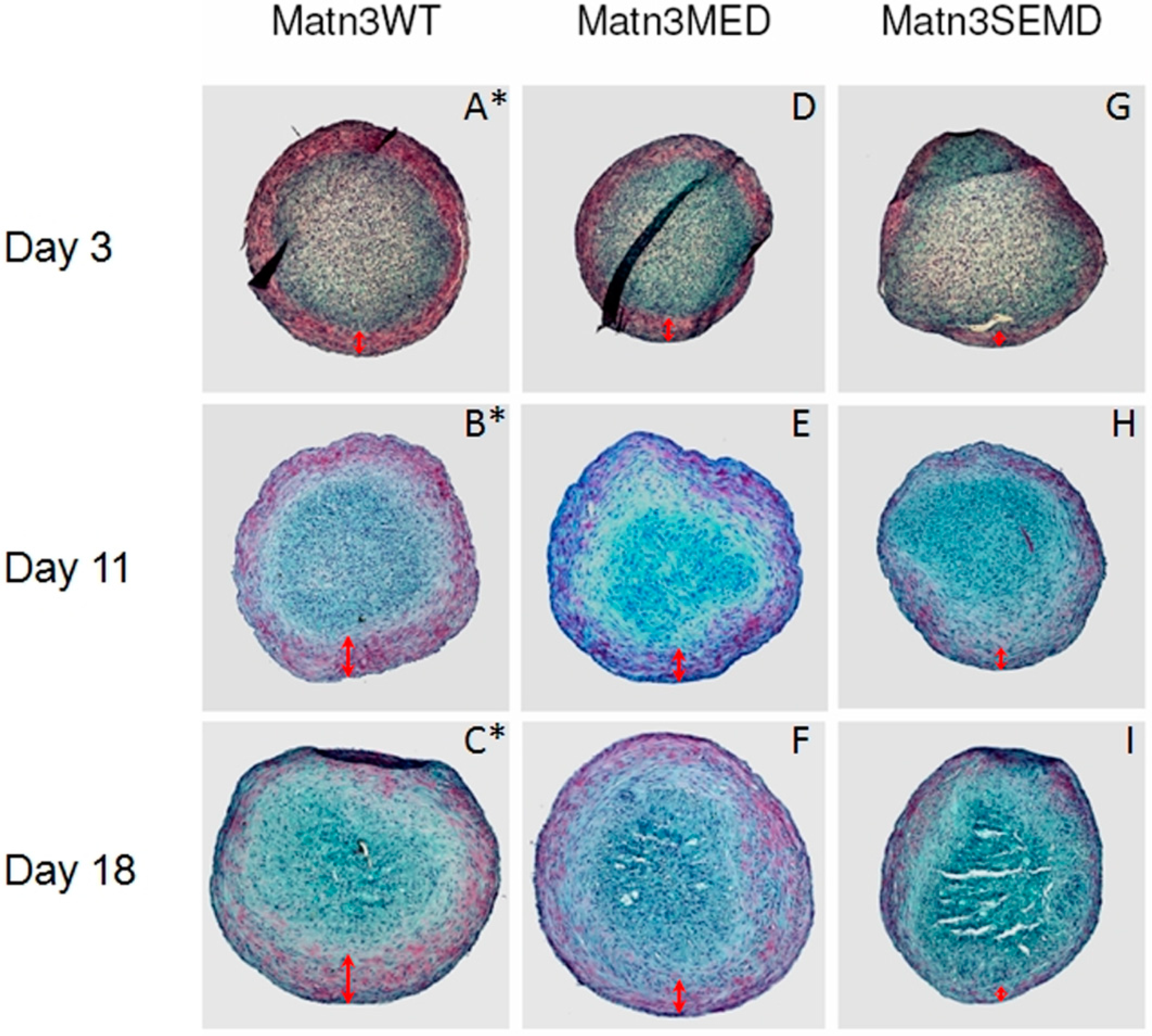
2.2. WT MATN3 Induces Spontaneous Chondrogenesis, while MED and SEMD Mutations Diminish MATN3 Induced Chondrogenic Effects, in Chondroprogenitors


2.3. TGF-β1 Is Incapable of Restoring Normal Chondrogenic Phenotype to Cells Expressing the MED or SEMD MATN3 Mutation

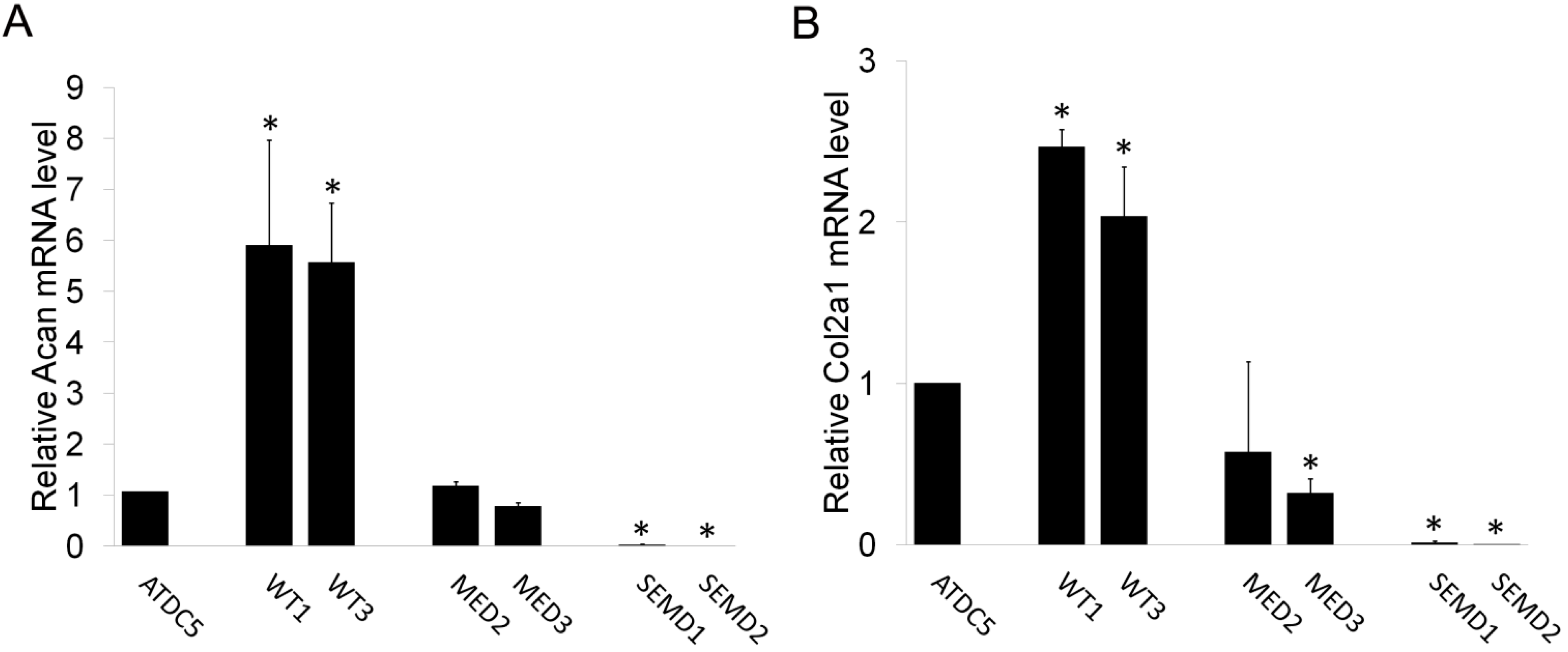
2.4. The Effect of WT and Mutant MATN3 on Chondrogenesis Is Consistent among Different Cell Lines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Generated Clones | MATN3 Expression Levels (Relative to Parental ATDC5) |
|---|---|---|
| WT MATN3 | WT1 | 2.6 |
| WT2 | 18.4 | |
| WT3 | 25.7 | |
| MED MATN3 | MED1 | 1.5 |
| MED2 | 1.9 | |
| MED3 | 17.2 | |
| SEMD MATN3 | SEMD1 | 6.9 |
| SEMD2 | 46.8 | |
| SEMD3 | 1122 |
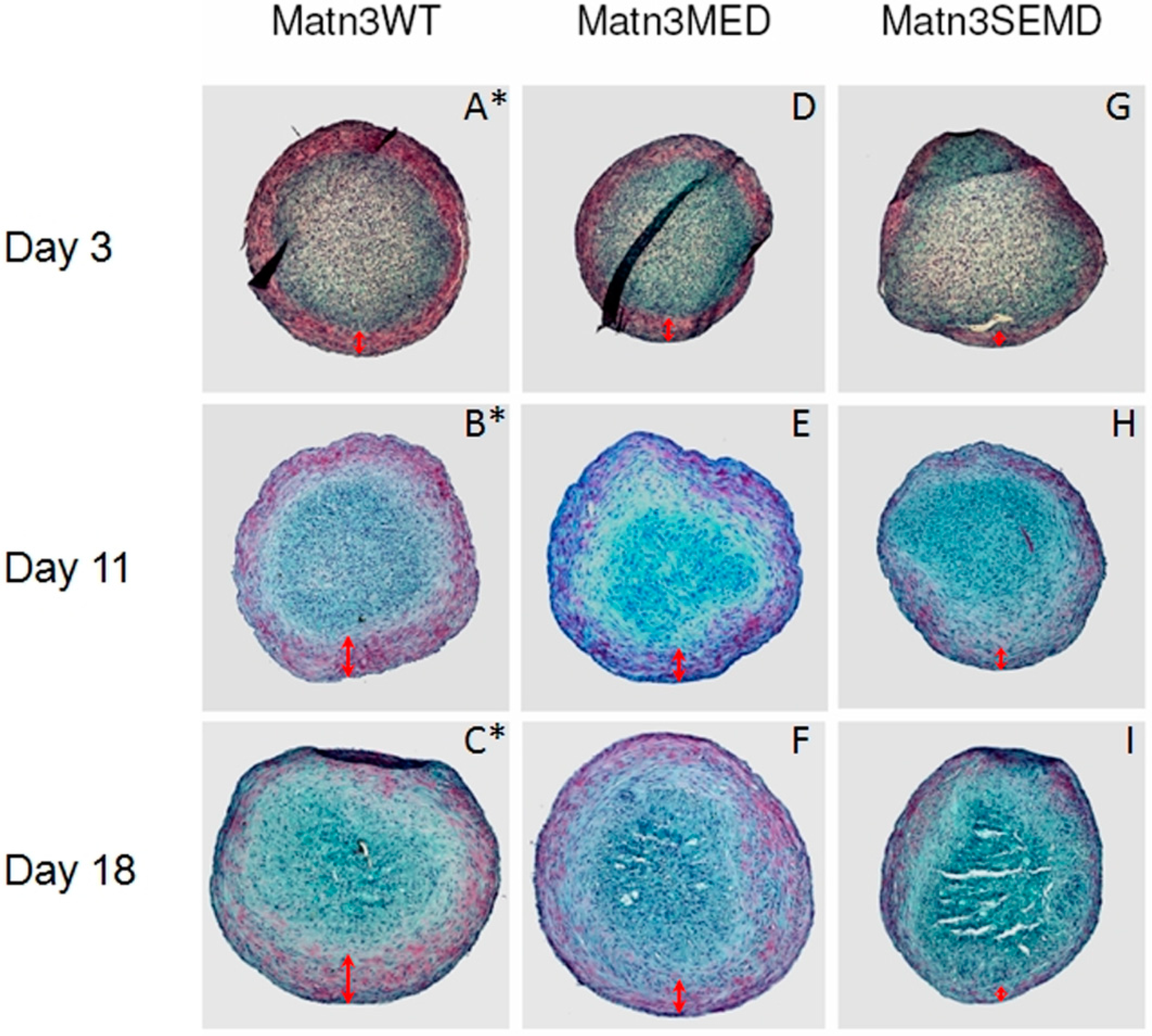
2.5. Synovium Derived Mesenchymal Stem Cells Expressing SEMD MATN3 Exhibit Diminished Chondrogenesis Potential

3. Experimental Section
3.1. Cloning and Construction of MATN3 cDNAs
3.2. Establishing ATDC5 Chondroprogenitor Cell Lines and SDMSCs Harboring MATN3 Mutations
3.3. SDS-Polyacrylamide Gel Electrophoresis and Western Blot Analysis
3.4. Experimental Cell Culture Conditions
3.5. RNA Isolation and Real-Time RT-PCR
| Gene | Forward | Reverse |
|---|---|---|
| Acan | CAGTGCGATGCAGGCTGGCT | CCTCCGGCACTCGTTGGCTG |
| Col2a1 | CACACTGGTAAGTGGGGCAAGACCG | GGATTGTGTTGTTTCAGGGTTCGGG |
| Col10a1 | GCCAGGAAAGCTGCCCCACG | GAGGTCCGGTTGGGCCTGGT |
| MATN3 | TTCCACCCGCGCGCCATATTC | CGTGTCTGTGGCCCCGATGTC |
3.6. Alcian Blue Staining and Quantification
3.7. Histology and Safranin-O Staining of Primary SDMSC Cell Pellets
3.8. GAG Quantification with Dimethylmethylene Blue Assay
3.9. Statistical Analysis
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chapman, K.L.; Mortier, G.R.; Chapman, K.; Loughlin, J.; Grant, M.E.; Briggs, M.D. Mutations in the Region Encoding the Von Willebrand Factor A Domain of Matrilin-3 Are Associated with Multiple Epiphyseal Dysplasia. Nat. Genet. 2001, 28, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Borochowitz, Z.U.; Scheffer, D.; Adir, V.; Dagoneau, N.; Munnich, A.; Cormier-Daire, V. Spondylo-Epi-Metaphyseal Dysplasia (SEMD) Matrilin 3 Type: Homozygote Matrilin 3 Mutation in a Novel Form of SEMD. J. Med. Genet. 2004, 41, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Klatt, A.R.; Nitsche, D.P.; Kobbe, B.; Morgelin, M.; Paulsson, M.; Wagener, R. Molecular Structure and Tissue Distribution of Matrilin-3, a Filament-Forming Extracellular Matrix Protein Expressed during Skeletal Development. J. Biol. Chem. 2000, 275, 3999–4006. [Google Scholar] [CrossRef] [PubMed]
- Deak, F.; Wagener, R.; Kiss, I.; Paulsson, M. The Matrilins: A Novel Family of Oligomeric Extracellular Matrix Proteins. Matrix Biol. 1999, 18, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Budde, B.; Blumbach, K.; Ylostalo, J.; Zaucke, F.; Ehlen, H.W.; Wagener, R.; Ala-Kokko, L.; Paulsson, M.; Bruckner, P.; Grassel, S. Altered Integration of Matrilin-3 into Cartilage Extracellular Matrix in the Absence of Collagen IX. Mol. Cell. Biol. 2005, 25, 10465–10478. [Google Scholar] [CrossRef] [PubMed]
- Fresquet, M.; Jowitt, T.A.; Ylostalo, J.; Coffey, P.; Meadows, R.S.; Ala-Kokko, L.; Thornton, D.J.; Briggs, M.D. Structural and Functional Characterization of Recombinant Matrilin-3 A-Domain and Implications for Human Genetic Bone Diseases. J. Biol. Chem. 2007, 282, 34634–34643. [Google Scholar] [CrossRef] [PubMed]
- Mann, H.H.; Ozbek, S.; Engel, J.; Paulsson, M.; Wagener, R. Interactions between the Cartilage Oligomeric Matrix Protein and Matrilins. Implications for Matrix Assembly and the Pathogenesis of Chondrodysplasias. J. Biol. Chem. 2004, 279, 25294–25298. [Google Scholar]
- Klatt, A.R.; Paulsson, M.; Wagener, R. Expression of Matrilins during Maturation of Mouse Skeletal Tissues. Matrix Biol. 2002, 21, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Eyre, D.R. Matrilin-3 Forms Disulfide-Linked Oligomers with Matrilin-1 in Bovine Epiphyseal Cartilage. J. Biol. Chem. 1998, 273, 17433–17438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Q. Changes of Matrilin Forms during Endochondral Ossification. Molecular Basis of Oligomeric Assembly. J. Biol. Chem. 2000, 275, 32628–32634. [Google Scholar]
- Treble, N.J.; Jensen, F.O.; Bankier, A.; Rogers, J.G.; Cole, W.G. Development of the Hip in Multiple Epiphyseal Dysplasia. Natural History and Susceptibility to Premature Osteoarthritis. J. Bone Jt. Surg. Br. 1990, 72, 1061–1064. [Google Scholar]
- Unger, S.L.; Briggs, M.D.; Holden, P.; Zabel, B.; Ala-Kokko, L.; Paassilta, P.; Lohiniva, J.; Rimoin, D.L.; Lachman, R.S.; Cohn, D.H. Multiple Epiphyseal Dysplasia: Radiographic Abnormalities Correlated with Genotype. Pediatr. Radiol. 2001, 31, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Makitie, O.; Mortier, G.R.; Czarny-Ratajczak, M.; Wright, M.J.; Suri, M.; Rogala, P.; Freund, M.; Jackson, G.C.; Jakkula, E.; Ala-Kokko, L.; et al. Clinical and Radiographic Findings in Multiple Epiphyseal Dysplasia Caused by MATN3 Mutations: Description of 12 Patients. Am. J. Med. Genet. A 2004, 125A, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.C.; Barker, F.S.; Jakkula, E.; Czarny-Ratajczak, M.; Makitie, O.; Cole, W.G.; Wright, M.J.; Smithson, S.F.; Suri, M.; Rogala, P.; et al. Missense Mutations in the Β Strands of the Single A-Domain of Matrilin-3 Result in Multiple Epiphyseal Dysplasia. J. Med. Genet. 2004, 41, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, A.; Haga, N.; Maeda, K.; Nakashima, E.; Manabe, N.; Hiraoka, H.; Kitoh, H.; Kosaki, R.; Nishimura, G.; Ohashi, H.; et al. Novel and Recurrent Mutations Clustered in the Von Willebrand Factor A Domain of MATN3 in Multiple Epiphyseal Dysplasia. Hum. Mutat. 2004, 24, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Otten, C.; Wagener, R.; Paulsson, M.; Zaucke, F. Matrilin-3 Mutations that Cause Chondrodysplasias Interfere with Protein Trafficking while a Mutation Associated with Hand Osteoarthritis does Not. J. Med. Genet. 2005, 42, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Cotterill, S.L.; Jackson, G.C.; Leighton, M.P.; Wagener, R.; Makitie, O.; Cole, W.G.; Briggs, M.D. Multiple Epiphyseal Dysplasia Mutations in MATN3 Cause Misfolding of the A-Domain and Prevent Secretion of Mutant Matrilin-3. Hum. Mutat. 2005, 26, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Fresquet, M.; Jackson, G.C.; Loughlin, J.; Briggs, M.D. Novel Mutations in Exon 2 of MATN3 Affect Residues within the Alpha-Helices of the A-Domain and can Result in the Intracellular Retention of Mutant Matrilin-3. Hum. Mutat. 2008, 29, 330. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.H.; Park, H.; Seong, M.W.; Cho, T.J.; Nishimura, G.; Superti-Furga, A.; Unger, S.; Ikegawa, S.; Choi, I.H.; Song, H.R.; et al. Revisit of Multiple Epiphyseal Dysplasia: Ethnic Difference in Genotypes and Comparison of Radiographic Features Linked to the COMP and MATN3 Genes. Am. J. Med. Genet. A 2011, 155A, 2669–2680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.K.; Luo, J.M.; Kanbe, K.; Chen, Q. Multiple Functions of the Von Willebrand Factor A Domain in Matrilins: Secretion, Assembly, and Proteolysis. J. Orthop. Surg. Res. 2008, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Leighton, M.P.; Nundlall, S.; Starborg, T.; Meadows, R.S.; Suleman, F.; Knowles, L.; Wagener, R.; Thornton, D.J.; Kadler, K.E.; Boot-Handford, R.P.; et al. Decreased Chondrocyte Proliferation and Dysregulated Apoptosis in the Cartilage Growth Plate are Key Features of a Murine Model of Epiphyseal Dysplasia Caused by a MATN3 Mutation. Hum. Mol. Genet. 2007, 16, 1728–1741. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wang, J.; Moore, D.C.; Liang, H.; Dooner, M.; Wu, Q.; Terek, R.; Chen, Q.; Ehrlich, M.G.; Quesenberry, P.J.; et al. Ptpn11 Deletion in a Novel Progenitor Causes Metachondromatosis by Inducing Hedgehog Signalling. Nature 2013, 499, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Johnson, D.M.; Haudenschild, D.R.; Goetinck, P.F. Progression and Recapitulation of the Chondrocyte Differentiation Program: Cartilage Matrix Protein is a Marker for Cartilage Maturation. Dev. Biol. 1995, 172, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Miwa, Y.; Kimata, K.; Ikawa, Y. A Chondrogenic Cell Line Derived from a Differentiating Culture of AT805 Teratocarcinoma Cells. Cell Differ. Dev. 1990, 30, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Nepal, M.; Park, Y.R.; Lee, H.K.; Oh, S.R.; Soh, Y. Stimulation of Chondrogenesis in ATDC5 Chondroprogenitor Cells and Hypertrophy in Mouse by Genkwadaphnin. Eur. J. Pharmacol. 2011, 655, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Pei, M.; Luo, J.; Chen, Q. Enhancing and Maintaining Chondrogenesis of Synovial Fibroblasts by Cartilage Extracellular Matrix Protein Matrilins. Osteoarthr. Cartil. 2008, 16, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Pei, M.; He, F.; Vunjak-Novakovic, G. Synovium-Derived Stem Cell-Based Chondrogenesis. Differentiation 2008, 76, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Bilgen, B.; Ren, Y.; Pei, M.; Aaron, R.K.; Ciombor, D.M. CD14-Negative Isolation Enhances Chondrogenesis in Synovial Fibroblasts. Tissue Eng. Part A 2009, 15, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Farndale, R.W.; Buttle, D.J.; Barrett, A.J. Improved Quantitation and Discrimination of Sulphated Glycosaminoglycans by use of Dimethylmethylene Blue. Biochim. Biophys. Acta 1986, 883, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Mostert, A.K.; Dijkstra, P.F.; Jansen, B.R.; van Horn, J.R.; de Graaf, B.; Heutink, P.; Lindhout, D. Familial Multiple Epiphyseal Dysplasia due to a Matrilin-3 Mutation: Further Delineation of the Phenotype Including 40 Years Follow-Up. Am. J. Med. Genet. A 2003, 120A, 490–497. [Google Scholar] [CrossRef] [PubMed]
- O'Sullivan, J.; D'Arcy, S.; Barry, F.P.; Murphy, J.M.; Coleman, C.M. Mesenchymal Chondroprogenitor Cell Origin and Therapeutic Potential. Stem Cell. Res. Ther. 2011, 2, 8. [Google Scholar]
- Dowthwaite, G.P.; Bishop, J.C.; Redman, S.N.; Khan, I.M.; Rooney, P.; Evans, D.J.; Haughton, L.; Bayram, Z.; Boyer, S.; Thomson, B.; et al. The Surface of Articular Cartilage Contains a Progenitor Cell Population. J. Cell. Sci. 2004, 117, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Barna, M.; Niswander, L. Visualization of Cartilage Formation: Insight into Cellular Properties of Skeletal Progenitors and Chondrodysplasia Syndromes. Dev. Cell. 2007, 12, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Zuscik, M.J.; Hilton, M.J.; Zhang, X.; Chen, D.; O’Keefe, R.J. Regulation of Chondrogenesis and Chondrocyte Differentiation by Stress. J. Clin. Investig. 2008, 118, 429–438. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Barnes, K.M.; Uyeda, J.A.; De-Levi, S.; Abad, V.; Palese, T.; Mericq, V.; Baron, J. Regulation of Growth Plate Chondrogenesis by Bone Morphogenetic Protein-2. Endocrinology 2001, 142, 430–436. [Google Scholar]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone Deacetylase 4 Controls Chondrocyte Hypertrophy during Skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Ballock, R.T.; Heydemann, A.; Wakefield, L.M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B. TGF-Β 1 Prevents Hypertrophy of Epiphyseal Chondrocytes: Regulation of Gene Expression for Cartilage Matrix Proteins and Metalloproteases. Dev. Biol. 1993, 158, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; Blom, A.; van den Berg, W.B. TGF-Β Signaling in Chondrocyte Terminal Differentiation and Osteoarthritis: Modulation and Integration of Signaling Pathways through Receptor-Smads. Osteoarthr. Cartil. 2009, 17, 1539–1545. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jayasuriya, C.T.; Zhou, F.H.; Pei, M.; Wang, Z.; Lemme, N.J.; Haines, P.; Chen, Q. Matrilin-3 Chondrodysplasia Mutations Cause Attenuated Chondrogenesis, Premature Hypertrophy and Aberrant Response to TGF-β in Chondroprogenitor Cells. Int. J. Mol. Sci. 2014, 15, 14555-14573. https://doi.org/10.3390/ijms150814555
Jayasuriya CT, Zhou FH, Pei M, Wang Z, Lemme NJ, Haines P, Chen Q. Matrilin-3 Chondrodysplasia Mutations Cause Attenuated Chondrogenesis, Premature Hypertrophy and Aberrant Response to TGF-β in Chondroprogenitor Cells. International Journal of Molecular Sciences. 2014; 15(8):14555-14573. https://doi.org/10.3390/ijms150814555
Chicago/Turabian StyleJayasuriya, Chathuraka T., Fiona H. Zhou, Ming Pei, Zhengke Wang, Nicholas J. Lemme, Paul Haines, and Qian Chen. 2014. "Matrilin-3 Chondrodysplasia Mutations Cause Attenuated Chondrogenesis, Premature Hypertrophy and Aberrant Response to TGF-β in Chondroprogenitor Cells" International Journal of Molecular Sciences 15, no. 8: 14555-14573. https://doi.org/10.3390/ijms150814555
APA StyleJayasuriya, C. T., Zhou, F. H., Pei, M., Wang, Z., Lemme, N. J., Haines, P., & Chen, Q. (2014). Matrilin-3 Chondrodysplasia Mutations Cause Attenuated Chondrogenesis, Premature Hypertrophy and Aberrant Response to TGF-β in Chondroprogenitor Cells. International Journal of Molecular Sciences, 15(8), 14555-14573. https://doi.org/10.3390/ijms150814555