Defining Molecular Sensors to Assess Long-Term Effects of Pesticides on Carcinogenesis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ligands and Agonists of Aryl Hydrocarbon Receptor (AhR)
3. Endogenous Ligands
4. Exogenous Ligands
4.1. “Classical” Synthetic AhR Ligands
4.2. Natural/Dietary Compounds
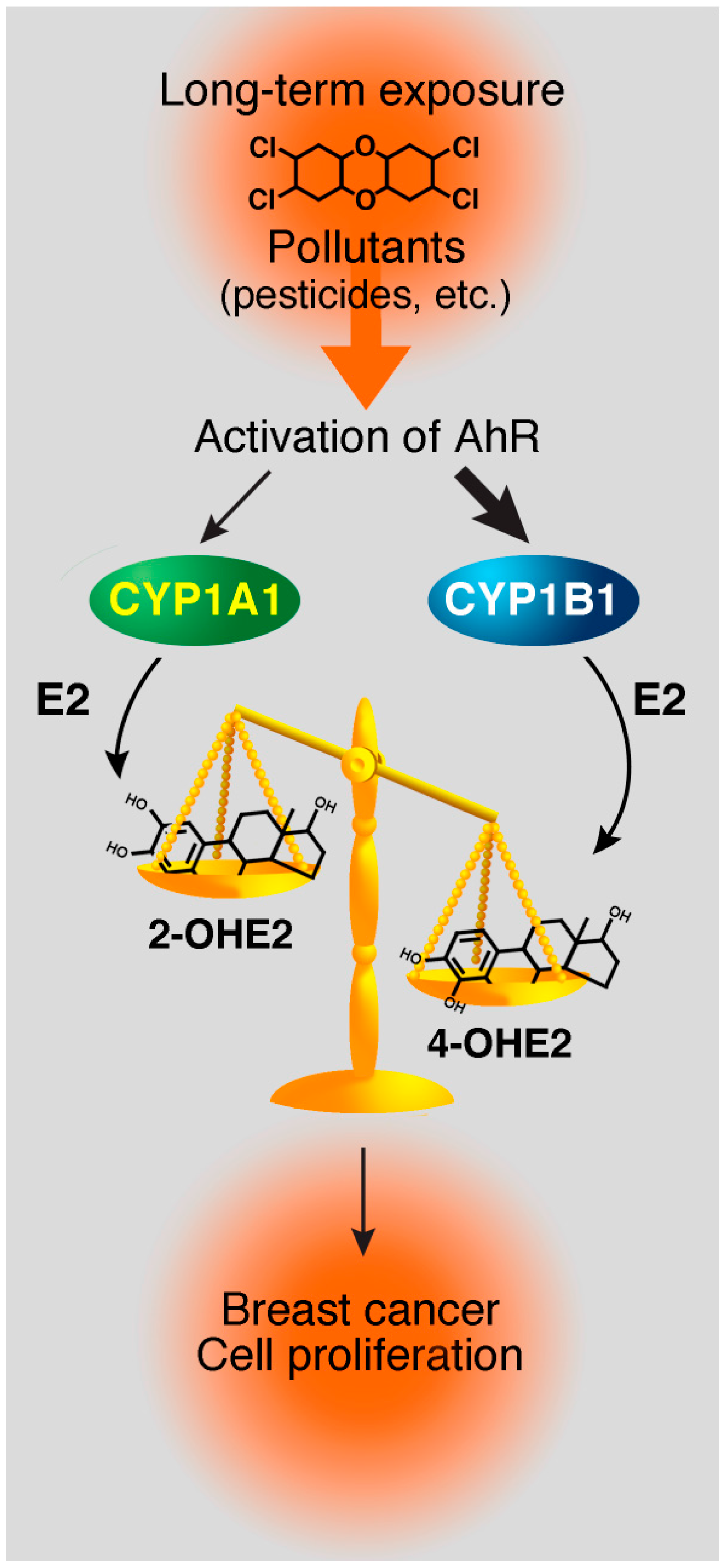
4.3. Pesticides

5. Molecular Mechanisms of AhR and ERα Crosstalk
6. AhR-Mediated Repression of the ERα Signaling Pathway
7. ERα-Mediated Repression of AhR Target Genes

8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hankinson, O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 307–340. [Google Scholar]
- Kazlauskas, A.; Sundstrom, S.; Poellinger, L.; Pongratz, I. The HSP90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol. Cell. Biol. 2001, 21, 2594–607. [Google Scholar]
- Nebert, D.W.; Gonzalez, F.J. P450 genes: Structure, evolution, and regulation. Annu. Rev. Biochem. 1987, 56, 945–993. [Google Scholar]
- Poland, A.; Glover, E. Chlorinated biphenyl induction of aryl hydrocarbon hydroxylase activity: A study of the structure-activity relationship. Mol. Pharmacol. 1977, 13, 924–938. [Google Scholar]
- Esser, C.; Rannug, A.; Stockinger, B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009, 30, 447–454. [Google Scholar]
- Linden, J.; Lensu, S.; Tuomisto, J.; Pohjanvirta, R. Dioxins, the aryl hydrocarbon receptor and the central regulation of energy balance. Front. Neuroendocrinol. 2010, 31, 452–478. [Google Scholar]
- Stevens, E.A.; Mezrich, J.D.; Bradfield, C.A. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 2009, 127, 299–311. [Google Scholar]
- McMillan, B.J.; Bradfield, C.A. The aryl hydrocarbon receptor sans xenobiotics: Endogenous function in genetic model systems. Mol. Pharmacol. 2007, 72, 487–498. [Google Scholar]
- Schmidt, J.V.; Su, G.H.; Reddy, J.K.; Simon, M.C.; Bradfield, C.A. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. USA 1996, 93, 6731–6736. [Google Scholar]
- Tian, Y.; Ke, S.; Denison, M.S.; Rabson, A.B.; Gallo, M.A. Ah receptor and NF-κB interactions, a potential mechanism for dioxin toxicity. J. Biol. Chem. 1999, 274, 510–515. [Google Scholar]
- Kim, D.W.; Gazourian, L.; Quadri, S.A.; Romieu-Mourez, R.; Sherr, D.H.; Sonenshein, G.E. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 2000, 19, 5498–506. [Google Scholar]
- Hayes, J.D.; Dinkova-Kostova, A.T.; McMahon, M. Cross-talk between transcription factors AhR and Nrf2: Lessons for cancer chemoprevention from dioxin. Toxicol. Sci. 2009, 111, 199–201. [Google Scholar]
- Puga, A.; Barnes, S.J.; Dalton, T.P.; Chang, C.; Knudsen, E.S.; Maier, M.A. Aromatic hydrocarbon receptor interaction with the retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J. Biol. Chem. 2000, 275, 2943–2950. [Google Scholar]
- Wang, F.; Wang, W.; Safe, S. Regulation of constitutive gene expression through interactions of Sp1 protein with the nuclear aryl hydrocarbon receptor complex. Biochemistry 1999, 38, 11490–11500. [Google Scholar]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar]
- Beischlag, T.V.; Perdew, G.H. ERα–AHR–ARNT protein–protein interactions mediate estradiol-dependent transrepression of dioxin-inducible gene transcription. J. Biol. Chem. 2005, 280, 21607–21611. [Google Scholar]
- Matthews, J.; Wihlen, B.; Thomsen, J.; Gustafsson, J.A. Aryl hydrocarbon receptor-mediated transcription: Ligand-dependent recruitment of estrogen receptor alpha to 2,3,7,8-tetrachlorodibenzo-p-dioxin-responsive promoters. Mol. Cell Biol. 2005, 25, 5317–5328. [Google Scholar]
- Ohtake, F.; Takeyama, K.; Matsumoto, T.; Kitagawa, H.; Yamamoto, Y.; Nohara, K.; Tohyama, C.; Krust, A.; Mimura, J.; Chambon, P.; et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 2003, 423, 545–550. [Google Scholar]
- Wormke, M.; Stoner, M.; Saville, B.; Walker, K.; Abdelrahim, M.; Burghardt, R.; Safe, S. The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes. Mol. Cell Biol. 2003, 23, 1843–1855. [Google Scholar]
- Powell, J.B.; Goode, G.D.; Eltom, S.E. The aryl hydrocarbon receptor: A target for breast cancer therapy. J. Cancer Ther. 2013, 4, 1177–1186. [Google Scholar]
- Yang, X.; Solomon, S.; Fraser, L.R.; Trombino, A.F.; Liu, D.; Sonenshein, G.E.; Hestermann, E.V.; Sherr, D.H. Constitutive regulation of CYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J. Cell. Biochem. 2008, 104, 402–417. [Google Scholar]
- Korzeniewski, N.; Wheeler, S.; Chatterjee, P.; Duensing, A.; Duensing, S. A novel role of the aryl hydrocarbon receptor (AhR) in centrosome amplification - implications for chemoprevention. Mol. Cancer 2010, 9, 153. [Google Scholar]
- Wirgin, I.; Roy, N.K.; Loftus, M.; Chambers, R.C.; Franks, D.G.; Hahn, M.E. Mechanistic basis of resistance to PCBs in Atlantic tomcod from the Hudson River. Science 2011, 331, 1322–1325. [Google Scholar]
- Stejskalova, L.; Dvorak, Z.; Pavek, P. Endogenous and exogenous ligands of aryl hydrocarbon receptor: Current state of art. Curr. Drug Metab. 2011, 12, 198–212. [Google Scholar]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar]
- Nguyen, L.P.; Bradfield, C.A. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008, 21, 102–116. [Google Scholar]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [Green Version]
- Marinkovic, N.; Pasalic, D.; Ferencak, G.; Grskovic, B.; Stavljenic Rukavina, A. Dioxins and human toxicity. Arh. Rada. Toksikol. 2010, 61, 445–453. [Google Scholar]
- Colborn, T.; vom Saal, F.S.; Soto, A.M. Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environ. Health Perspect. 1993, 101, 378–384. [Google Scholar]
- Welch, R.M.; Levin, W.; Conney, A.H. Estrogenic action of DDT and its analogs. Toxicol. Appl. Pharmacol. 1969, 14, 358–367. [Google Scholar]
- Bulger, W.H.; Muccitelli, R.M.; Kupfer, D. Studies on the in vivo and in vitro estrogenic activities of methoxychlor and its metabolites. Role of hepatic mono-oxygenase in methoxychlor activation. Biochem. Pharmacol. 1978, 27, 2417–2423. [Google Scholar]
- Cummings, A.M. Methoxychlor as a model for environmental estrogens. Crit. Rev. Toxicol. 1997, 27, 367–379. [Google Scholar]
- Coosen, R.; van Velsen, F.L. Effects of the β-isomer of hexachlorocyclohexane on estrogen-sensitive human mammary tumor cells. Toxicol. Appl. Pharmacol. 1989, 101, 310–318. [Google Scholar]
- Soto, A.M.; Sonnenschein, C.; Chung, K.L.; Fernandez, M.F.; Olea, N.; Serrano, F.O. The E-SCREEN assay as a tool to identify estrogens: An update on estrogenic environmental pollutants. Environ. Health Perspect. 1995, 103, 113–122. [Google Scholar]
- Garey, J.; Wolff, M.S. Estrogenic and antiprogestagenic activities of pyrethroid insecticides. Biochem. Biophys. Res. Commun. 1998, 251, 855–859. [Google Scholar]
- Anderson, H.R.; Vinggaard, A.M.; Rasmussenn, T.H.; Gjermandsen, I.M.; Bonefeld-Jorgensen, E.C. Effects of currently used pesticides in assays for estrogenicity, andrenogenicity, and aromatase activity in vitro. Toxicol. Appl. Pharmacol. 2002, 179, 1–12. [Google Scholar]
- Takeuchi, S.; Iida, M.; Yabushita, H.; Matsuda, T.; Kojima, H. In vitro screening for aryl hydrocarbon receptor agonistic activity in 200 pesticides using a highly sensitive reporter cell line, DR-EcoScreen cells, and in vivo mouse liver cytochrome P450–1A induction by propanil, diuron and linuron. Chemosphere 2008, 74, 155–165. [Google Scholar]
- Kojima, H.; Katsura, E.; Takeuchi, S.; Niiyama, K.; Kobayashi, K. Screening for estrogen and androgen receptor activities in 200 pesticides by in vitro reporter gene assays using Chinese hamster ovary cells. Environ. Health Perspect. 2004, 112, 524–531. [Google Scholar]
- Cassidy, R.A.; Natarajan, S.; Vaughan, G.M. The link between the insecticide heptachlor epoxide, estradiol, and breast cancer. Breast Cancer Res. Treat. 2005, 90, 55–64. [Google Scholar]
- Ibarluzea Jm, J.; Fernandez, M.F.; Santa-Marina, L.; Olea-Serrano, M.F.; Rivas, A.M.; Aurrekoetxea, J.J.; Exposito, J.; Lorenzo, M.; Torne, P.; Villalobos, M.; et al. Breast cancer risk and the combined effect of environmental estrogens. Cancer Causes Control 2004, 15, 591–600. [Google Scholar]
- Martucci, C.P.; Fishman, J. P450 enzymes of estrogen metabolism. Pharmacol. Ther. 1993, 57, 237–257. [Google Scholar]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar]
- Yager, J.D.; Liehr, J.G. Molecular mechanisms of estrogen carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 203–232. [Google Scholar]
- Newbold, R.R.; Liehr, J.G. Induction of uterine adenocarcinoma in CD-1 mice by catechol estrogens. Cancer Res. 2000, 60, 235–237. [Google Scholar]
- Liehr, J.G.; Fang, W.F.; Sirbasku, D.A.; Ari-Ulubelen, A. Carcinogenicity of catechol estrogens in Syrian hamsters. J. Steroid Biochem. 1986, 24, 353–356. [Google Scholar]
- Gupta, M.; McDougal, A.; Safe, S. Estrogenic and antiestrogenic activities of 16α- and 2-hydroxy metabolites of 17β-estradiol in MCF-7 and T47D human breast cancer cells. J. Steroid Biochem. Mol. Biol. 1998, 67, 413–419. [Google Scholar]
- Hurh, Y.J.; Chen, Z.H.; Na, H.K.; Han, S.Y.; Surh, Y.J. 2-Hydroxyestradiol induces oxidative DNA damage and apoptosis in human mammary epithelial cells. J. Toxicol. Environ. Health A 2004, 67, 1939–1953. [Google Scholar]
- Zhao, Z.; Kosinska, W.; Khmelnitsky, M.; Cavalieri, E.L.; Rogan, E.G.; Chakravarti, D.; Sacks, P.G.; Guttenplan, J.B. Mutagenic activity of 4-hydroxyestradiol, but not 2-hydroxyestradiol, in BB rat2 embryonic cells, and the mutational spectrum of 4-hydroxyestradiol. Chem. Res. Toxicol 2006, 19, 475–479. [Google Scholar]
- Coumoul, X.; Diry, M.; Robillot, C.; Barouki, R. Differential regulation of cytochrome P450 1A1 and 1B1 by a combination of dioxin and pesticides in the breast tumor cell line MCF-7. Cancer Res. 2001, 61, 3942–3948. [Google Scholar]
- Rogan, E.G.; Badawi, A.F.; Devanesan, P.D.; Meza, J.L.; Edney, J.A.; West, W.W.; Higginbotham, S.M.; Cavalieri, E.L. Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: Potential biomarkers of susceptibility to cancer. Carcinogenesis 2003, 24, 697–702. [Google Scholar]
- Stone, R. Epidemiology. Agent orange’s bitter harvest. Science 2007, 315, 176–179. [Google Scholar]
- The Health and Environment Linkages Initiative (HELI). Available online: http://www.who.int/heli/risks/toxics/chemicals/en/index.html (accessed on 23 September 2014).
- Johansson, I.; Ingelman-Sundberg, M. Genetic polymorphism and toxicology—With emphasis on cytochrome p450. Toxicol. Sci. 2011, 120, 1–13. [Google Scholar]
- Hewitt, S.C.; Korach, K.S. Estrogen receptors: Structure, mechanisms and function. Rev. Endocr. Metab. Disord. 2002, 3, 193–200. [Google Scholar]
- Kociba, R.J.; Keyes, D.G.; Beyer, J.E.; Carreon, R.M.; Wade, C.E.; Dittenber, D.A.; Kalnins, R.P.; Frauson, L.E.; Park, C.N.; Barnard, S.D.; et al. Results of a two-year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Toxicol. Appl. Pharmacol. 1978, 46, 279–303. [Google Scholar]
- Gierthy, J.F.; Lincoln, D.W., 2nd. Inhibition of postconfluent focus production in cultures of MCF-7 human breast cancer cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Breast Cancer Res. Treat. 1988, 12, 227–233. [Google Scholar]
- Gierthy, J.F.; Lincoln, D.W.; Gillespie, M.B.; Seeger, J.I.; Martinez, H.L.; Dickerman, H.W.; Kumar, S.A. Suppression of estrogen-regulated extracellular tissue plasminogen activator activity of MCF-7 cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cancer Res. 1987, 47, 6198–6203. [Google Scholar]
- Chen, I.; McDougal, A.; Wang, F.; Safe, S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis 1998, 19, 1631–1639. [Google Scholar]
- Leong, H.; Riby, J.E.; Firestone, G.L.; Bjeldanes, L.F. Potent ligand-independent estrogen receptor activation by 3,3'-diindolylmethane is mediated by cross talk between the protein kinase A and mitogen-activated protein kinase signaling pathways. Mol. Endocrinol. 2004, 18, 291–302. [Google Scholar]
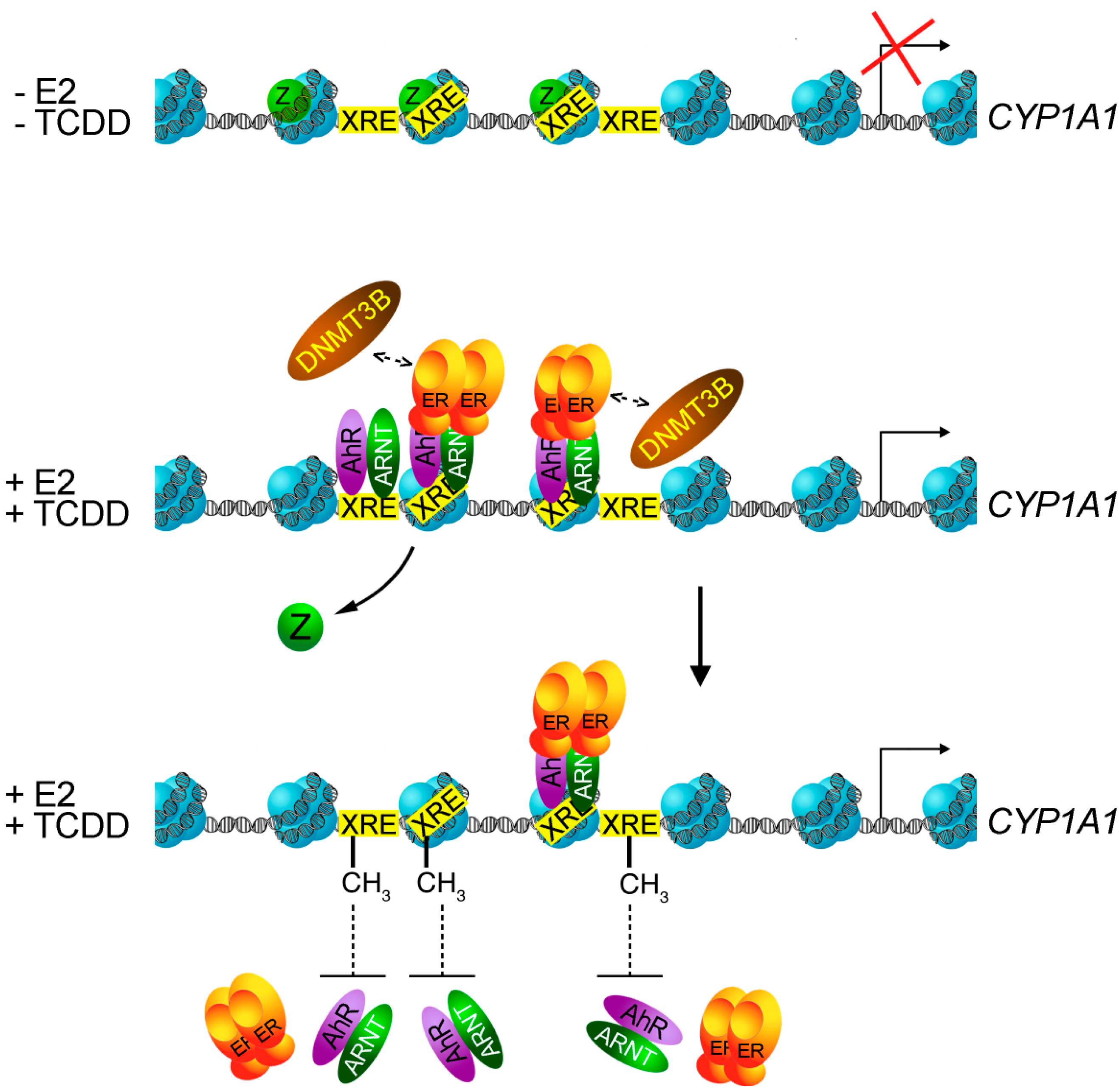
- Marques, M.; Laflamme, L.; Gaudreau, L. Estrogen receptor alpha can selectively repress dioxin receptor-mediated gene expression by targeting DNA methylation. Nucleic Acids Res. 2013, 41, 8094–8106. [Google Scholar]
- Nguyen, T.A.; Hoivik, D.; Lee, J.E.; Safe, S. Interactions of nuclear receptor coactivator/corepressor proteins with the aryl hydrocarbon receptor complex. Arch. Biochem. Biophys. 1999, 367, 250–257. [Google Scholar]
- Kumar, M.B.; Perdew, G.H. Nuclear receptor coactivator SRC-1 interacts with the Q-rich subdomain of the AhR and modulates its transactivation potential. Gene Expr. 1999, 8, 273–286. [Google Scholar]
- Kumar, M.B.; Tarpey, R.W.; Perdew, G.H. Differential recruitment of coactivator RIP140 by Ah and estrogen receptors. Absence of a role for LXXLL motifs. J. Biol. Chem. 1999, 274, 22155–22164. [Google Scholar]
- Kobayashi, A.; Numayama-Tsuruta, K.; Sogawa, K.; Fujii-Kuriyama, Y. CBP/p300 functions as a possible transcriptional coactivator of Ah receptor nuclear translocator (Arnt). J. Biochem. 1997, 122, 703–710. [Google Scholar]
- Krishnan, V.; Porter, W.; Santostefano, M.; Wang, X.; Safe, S. Molecular mechanism of inhibition of estrogen-induced cathepsin D gene expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in MCF-7 cells. Mol. Cell Biol. 1995, 15, 6710–6719. [Google Scholar]
- Wang, F.; Samudio, I.; Safe, S. Transcriptional activation of cathepsin D gene expression by 17β-estradiol: Mechanism of aryl hydrocarbon receptor-mediated inhibition. Mol. Cell Endocrinol. 2001, 172, 91–103. [Google Scholar]
- Duan, R.; Porter, W.; Samudio, I.; Vyhlidal, C.; Kladde, M.; Safe, S. Transcriptional activation of c-fos protooncogene by 17β-estradiol: Mechanism of aryl hydrocarbon receptor-mediated inhibition. Mol. Endocrinol. 1999, 13, 1511–1521. [Google Scholar]
- Porter, W.; Wang, F.; Duan, R.; Qin, C.; Castro-Rivera, E.; Kim, K.; Safe, S. Transcriptional activation of heat shock protein 27 gene expression by 17β-estradiol and modulation by antiestrogens and aryl hydrocarbon receptor agonists. J. Mol. Endocrinol. 2001, 26, 31–42. [Google Scholar]
- Gillesby, B.E.; Stanostefano, M.; Porter, W.; Safe, S.; Wu, Z.F.; Zacharewski, T.R. Identification of a motif within the 5' regulatory region of pS2 which is responsible for AP-1 binding and TCDD-mediated suppression. Biochemistry 1997, 36, 6080–6089. [Google Scholar]
- Harris, M.; Zacharewski, T.; Safe, S. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds on the occupied nuclear estrogen receptor in MCF-7 human breast cancer cells. Cancer Res. 1990, 50, 3579–3584. [Google Scholar]
- Lonard, D.M.; Nawaz, Z.; Smith, C.L.; O’Malley, B.W. The 26S proteasome is required for estrogen receptor-α and coactivator turnover and for efficient estrogen receptor-α transactivation. Mol. Cell 2000, 5, 939–948. [Google Scholar]
- Reid, G.; Hubner, M.R.; Metivier, R.; Brand, H.; Denger, S.; Manu, D.; Beaudouin, J.; Ellenberg, J.; Gannon, F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 2003, 11, 695–707. [Google Scholar]
- Shiverick, K.T.; Muther, T.F. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on serum concentrations and the uterotrophic action of exogenous estrone in rats. Toxicol. Appl. Pharmacol. 1982, 65, 170–176. [Google Scholar]
- Zilberman, D.; Coleman-Derr, D.; Ballinger, T.; Henikoff, S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 2008, 456, 125–129. [Google Scholar]
- Conerly, M.L.; Teves, S.S.; Diolaiti, D.; Ulrich, M.; Eisenman, R.N.; Henikoff, S. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res. 2010, 20, 1383–1390. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
L'Héritier, F.; Marques, M.; Fauteux, M.; Gaudreau, L. Defining Molecular Sensors to Assess Long-Term Effects of Pesticides on Carcinogenesis. Int. J. Mol. Sci. 2014, 15, 17148-17161. https://doi.org/10.3390/ijms150917148
L'Héritier F, Marques M, Fauteux M, Gaudreau L. Defining Molecular Sensors to Assess Long-Term Effects of Pesticides on Carcinogenesis. International Journal of Molecular Sciences. 2014; 15(9):17148-17161. https://doi.org/10.3390/ijms150917148
Chicago/Turabian StyleL'Héritier, Fanny, Maud Marques, Myriam Fauteux, and Luc Gaudreau. 2014. "Defining Molecular Sensors to Assess Long-Term Effects of Pesticides on Carcinogenesis" International Journal of Molecular Sciences 15, no. 9: 17148-17161. https://doi.org/10.3390/ijms150917148