
Trans-Splicing Improvement by the Combined Application of Antisense Strategies

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
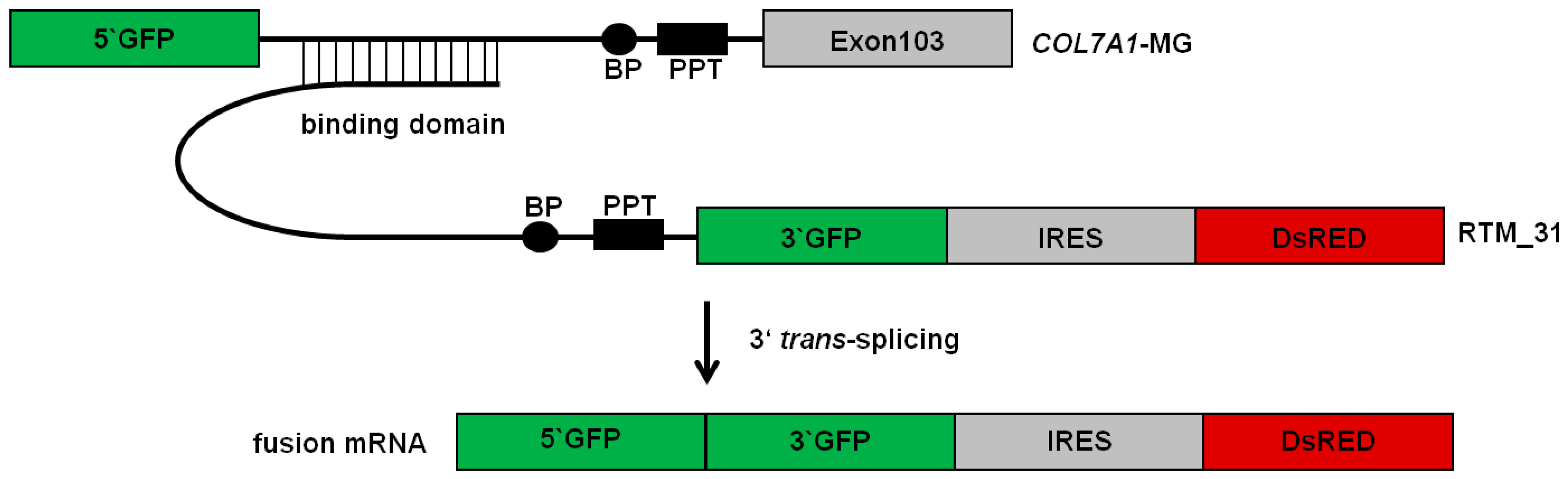
2.1. Selection of an Efficient Binding Domain Specific for Intron 102 of COL7A1

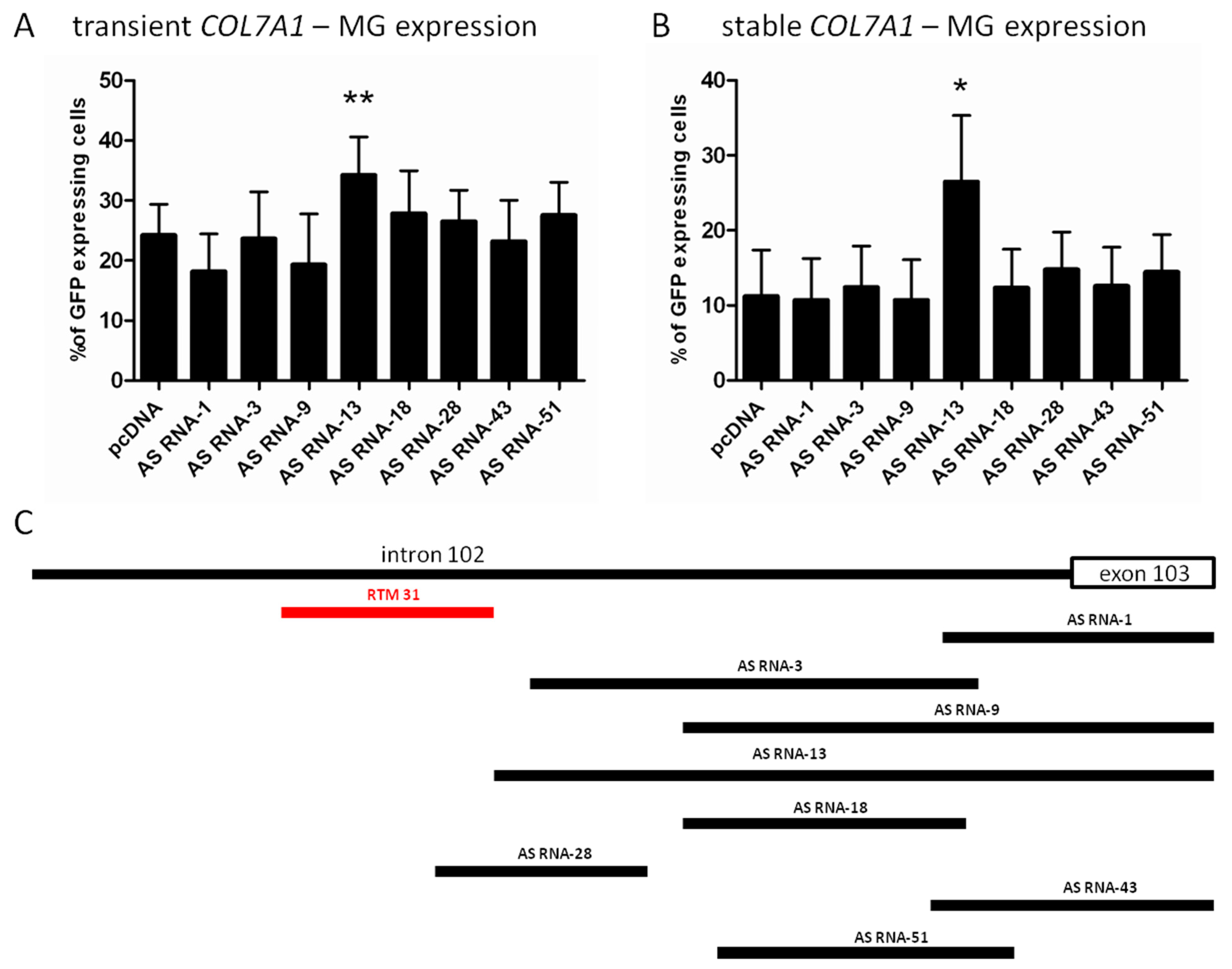
2.2. Selection of Antisense RNA Molecules (AS RNAs)
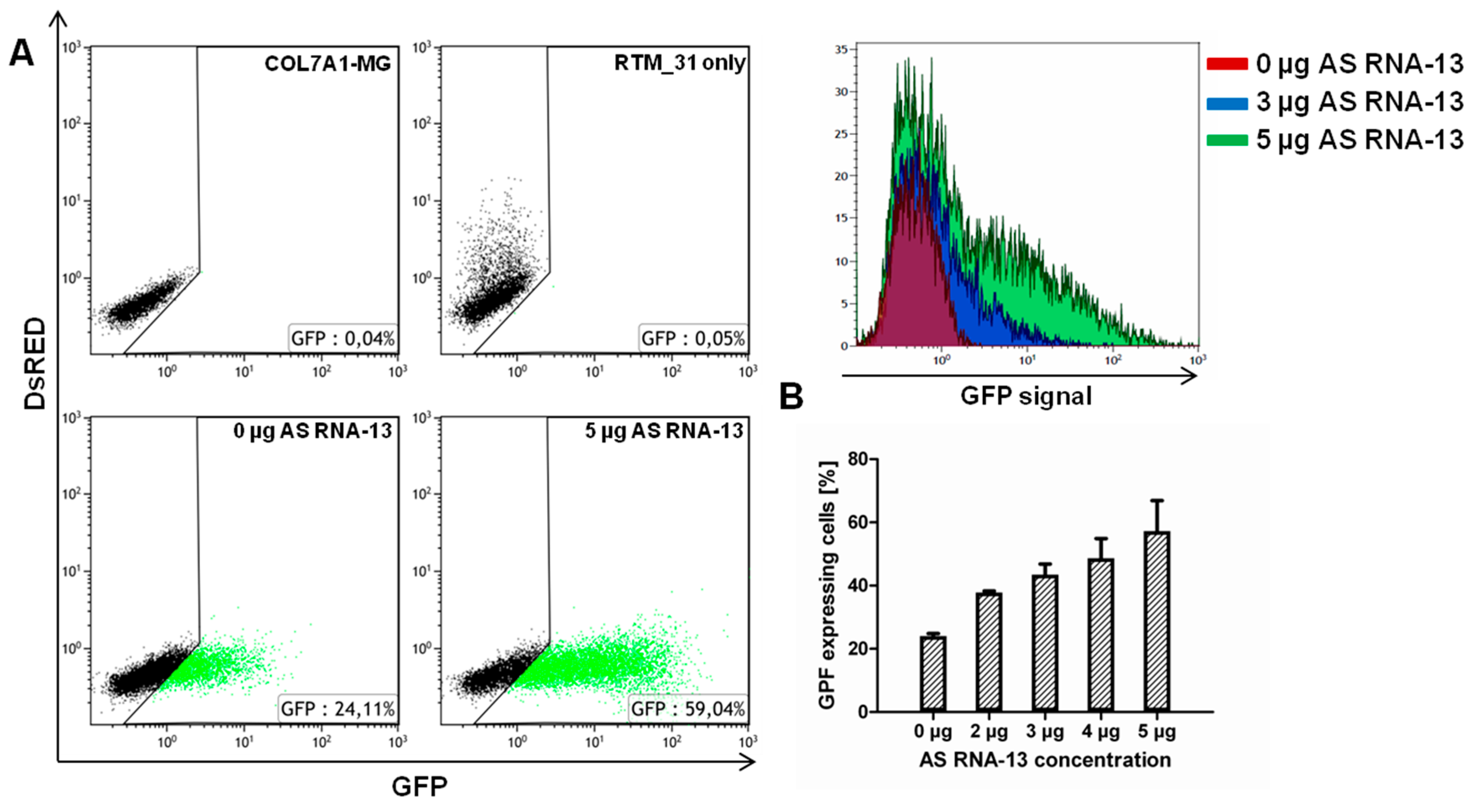
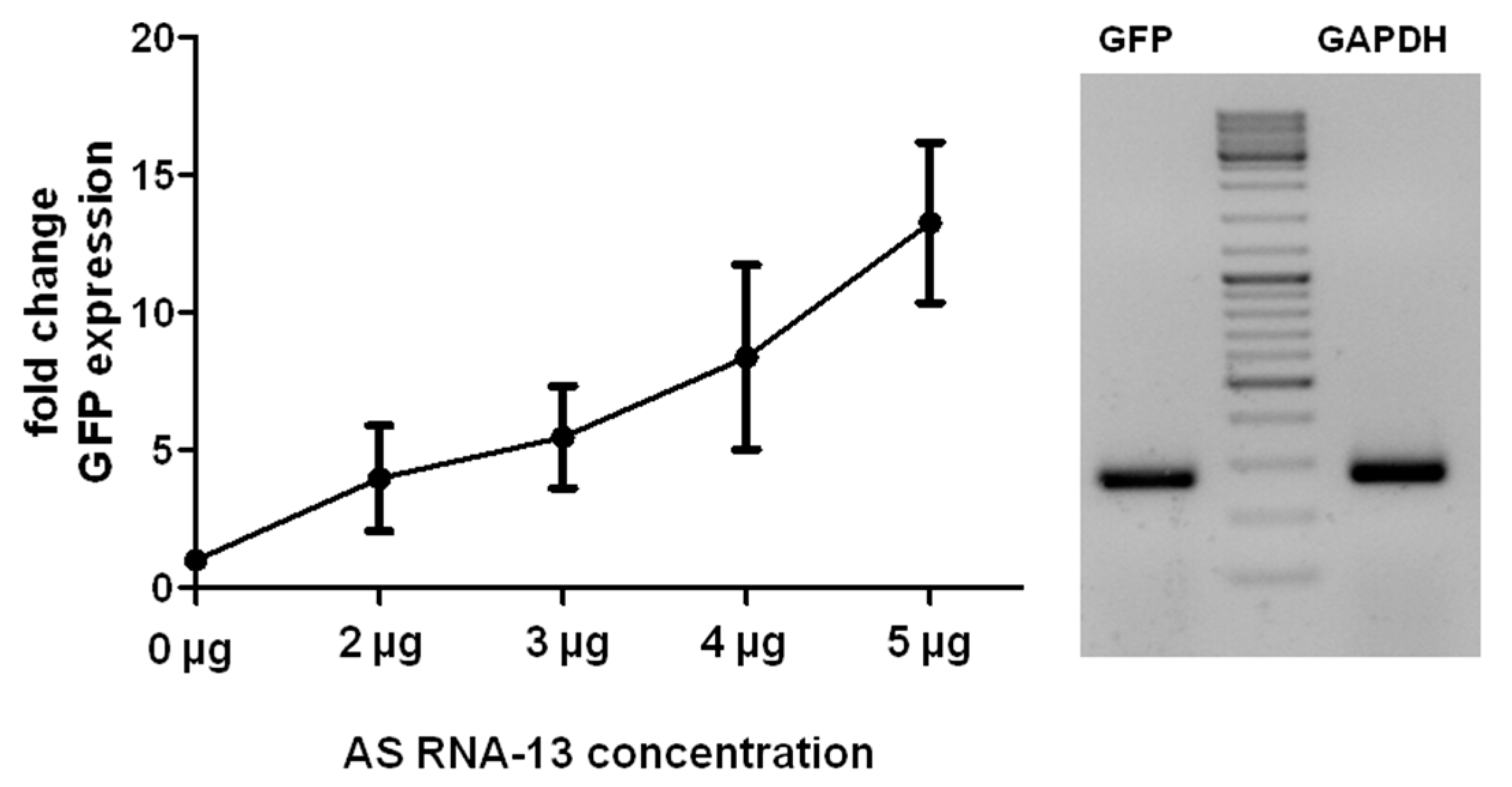
2.3. AS RNA-13 Improves Trans-Splicing Efficiency in a Dose-Dependent Manner


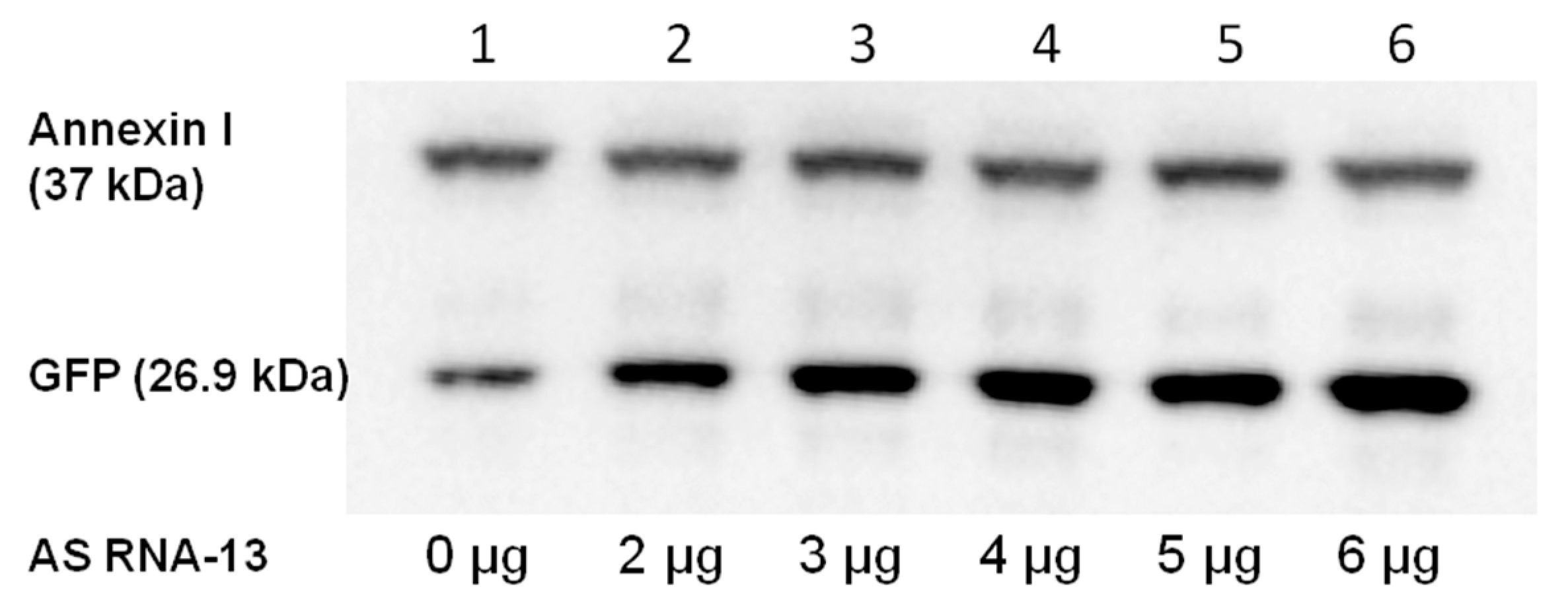
2.4. Detection of Full-Length GFP by Western Blot Analysis
2.5. Trans-Splicing Efficiency Analysis by Semiquantitative Real Time PCR (sqRT-PCR)


3. Discussion
4. Materials and Methods
4.1. Construction of Screening Plasmids
4.2. Cloning of COL7A1-Minigene PiggyBac (PB) Transposon Cassette
4.3. Development of Stable COL7A1-Minigene Expressing HEK293 Cell Line
4.4. Construction of Antisense Oligonucleotide (AS RNA) Library
4.5. Cell Culture and Plasmid Transfection
4.6. RNA Isolation and cDNA Synthesis
4.7. SqRT-PCR
4.8. Flow Cytometric Analysis
4.9. Western Blot Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chao, H.; Mansfield, S.G.; Bartel, R.C.; Hiriyanna, S.; Mitchell, L.G.; Garcia-Blanco, M.A.; Walsh, C.E. Phenotype correction of hemophilia A mice by spliceosome-mediated RNA trans-splicing. Nat. Med. 2003, 9, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, S.G.; Clark, R.H.; Puttaraju, M.; Kole, J.; Cohn, J.A.; Mitchell, L.G.; Garcia-Blanco, M.A. 5' Exon replacement and repair by spliceosome-mediated RNA trans-splicing. RNA 2003, 9, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Klausegger, A.; Koller, U.; Lochmuller, H.; Krause, S.; Wiche, G.; Mitchell, L.G.; Hintner, H.; Bauer, J.W. 5' Trans-splicing repair of the PLEC1 gene. J. Investig. Dermatol. 2008, 128, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Brunner, M.; Lettner, T.; Wagner, M.; Koller, U.; Trost, A.; Murauer, E.M.; Hainzl, S.; Hintner, H.; Bauer, J.W. K14 mRNA reprogramming for dominant epidermolysis bullosa simplex. Hum. Mol. Genet. 2010, 19, 4715–4725. [Google Scholar] [CrossRef] [PubMed]
- Murauer, E.M.; Gache, Y.; Gratz, I.K.; Klausegger, A.; Muss, W.; Gruber, C.; Meneguzzi, G.; Hintner, H.; Bauer, J.W. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2011, 131, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Murauer, E.M.; Bauer, J.W. Spliceosome-mediated trans-splicing: The therapeutic cut and paste. J. Investig. Dermatol. 2012, 132, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Titeux, M.; Pendaries, V.; Hovnanian, A. Gene therapy for recessive dystrophic epidermolysis bullosa. Dermatol. Clin. 2010, 28, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Koller, U.; Wally, V.; Mitchell, L.G.; Klausegger, A.; Murauer, E.M.; Mayr, E.; Gruber, C.; Hainzl, S.; Hintner, H.; Bauer, J.W. A novel screening system improves genetic correction by internal exon replacement. Nucleic Acids Res. 2011, 39, e108. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Koller, U.; Bauer, J.W. Human Genetic Diseases; Plaseska-Karanfilska, Dijana, Ed.; InTech: Shanghai, China, 2011; pp. 223–240. [Google Scholar]
- Murauer, E.M.; Koller, U.; Hainzl, S.; Wally, V.; Bauer, J.W. A reporter-based screen to identify potent 3' trans-splicing molecules for endogenous RNA repair. Hum. Gene Ther. Methods 2013, 24, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Koller, U.; Wally, V.; Bauer, J.W.; Murauer, E.M. Considerations for a successful RNA trans-splicing repair of genetic disorders. Mol. Ther. Nucleic Acids 2014, 3, e157. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.W.; Murauer, E.M.; Wally, V.; Koller, U. RNA trans-splicing for genodermatoses. Methods Mol. Biol. 2013, 961, 441–455. [Google Scholar] [PubMed]
- Coady, T.H.; Baughan, T.D.; Shababi, M.; Passini, M.A.; Lorson, C.L. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS One 2008, 3, e3468. [Google Scholar] [CrossRef] [PubMed]
- Coady, T.H.; Lorson, C.L. Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J. Neurosci. 2010, 30, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Hintner, H. Life with Epidermolysis Bullosa; Springer Wien: Wien, Austria, 2008. [Google Scholar]
- Fine, J.D.; Bruckner-Tuderman, L.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.; Gratz, I.K.; Murauer, E.M.; Mayr, E.; Koller, U.; Bruckner-Tuderman, L.; Meneguzzi, G.; Hintner, H.; Bauer, J.W. Spliceosome-mediated RNA trans-splicing facilitates targeted delivery of suicide genes to cancer cells. Mol. Cancer Ther. 2011, 10, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.; Koller, U.; Murauer, E.M.; Hainzl, S.; Hüttner, C.; Kocher, T.; South, A.P.; Hintner, H.; Bauer, J.W. The design and optimization of RNA trans-splicing molecules for skin cancer therapy. Mol. Oncol. 2013, 7, 1056–1068. [Google Scholar] [CrossRef]
- Puttaraju, M.; Jamison, S.F.; Mansfield, S.G.; Garcia-Blanco, M.A.; Mitchell, L.G. Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat. Biotechnol. 1999, 17, 246–225. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.G.; McGarrity, G.J. Gene therapy progress and prospects: Reprograming gene expression by trans-splicing. Gene Ther. 2005, 12, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Tidman, M.J.; Eady, R.A. Evaluation of anchoring fibrils and other components of the dermal-epidermal junction in dystrophic epidermolysis bullosa by a quantitative ultrastructural technique. J. Investig. Dermatol. 1985, 84, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, A.; Loeckermann, S.; Kern, J.S.; Braun, A.; Bosl, M.R.; Bley, T.A.; Schumann, H.; von Elverfeldt, D.; Paul, D.; Erlacher, M.; et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J. Clin. Investig. 2008, 118, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, A.; Spassov, S.; Elfert, S.; Schlosser, A.; Gache, Y.; Meneguzzi, G.; Bruckner-Tuderman, L. Dominant negative effects of COL7A1 mutations can be rescued by controlled over-expression of normal collagen VII. J. Biol. Chem. 2009, 284, 30248–30256. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.Y.; Longley, M.A.; Wang, X.J.; Roop, D.R. An inducible mouse model for epidermolysis bullosa simplex: Implications for gene therapy. J. Cell Biol. 2001, 152, 651–656. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koller, U.; Hainzl, S.; Kocher, T.; Hüttner, C.; Klausegger, A.; Gruber, C.; Mayr, E.; Wally, V.; Bauer, J.W.; Murauer, E.M. Trans-Splicing Improvement by the Combined Application of Antisense Strategies. Int. J. Mol. Sci. 2015, 16, 1179-1191. https://doi.org/10.3390/ijms16011179
Koller U, Hainzl S, Kocher T, Hüttner C, Klausegger A, Gruber C, Mayr E, Wally V, Bauer JW, Murauer EM. Trans-Splicing Improvement by the Combined Application of Antisense Strategies. International Journal of Molecular Sciences. 2015; 16(1):1179-1191. https://doi.org/10.3390/ijms16011179
Chicago/Turabian StyleKoller, Ulrich, Stefan Hainzl, Thomas Kocher, Clemens Hüttner, Alfred Klausegger, Christina Gruber, Elisabeth Mayr, Verena Wally, Johann W. Bauer, and Eva M. Murauer. 2015. "Trans-Splicing Improvement by the Combined Application of Antisense Strategies" International Journal of Molecular Sciences 16, no. 1: 1179-1191. https://doi.org/10.3390/ijms16011179