
Mechanisms of Hyperhomocysteinemia Induced Skeletal Muscle Myopathy after Ischemia in the CBS−/+ Mouse Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
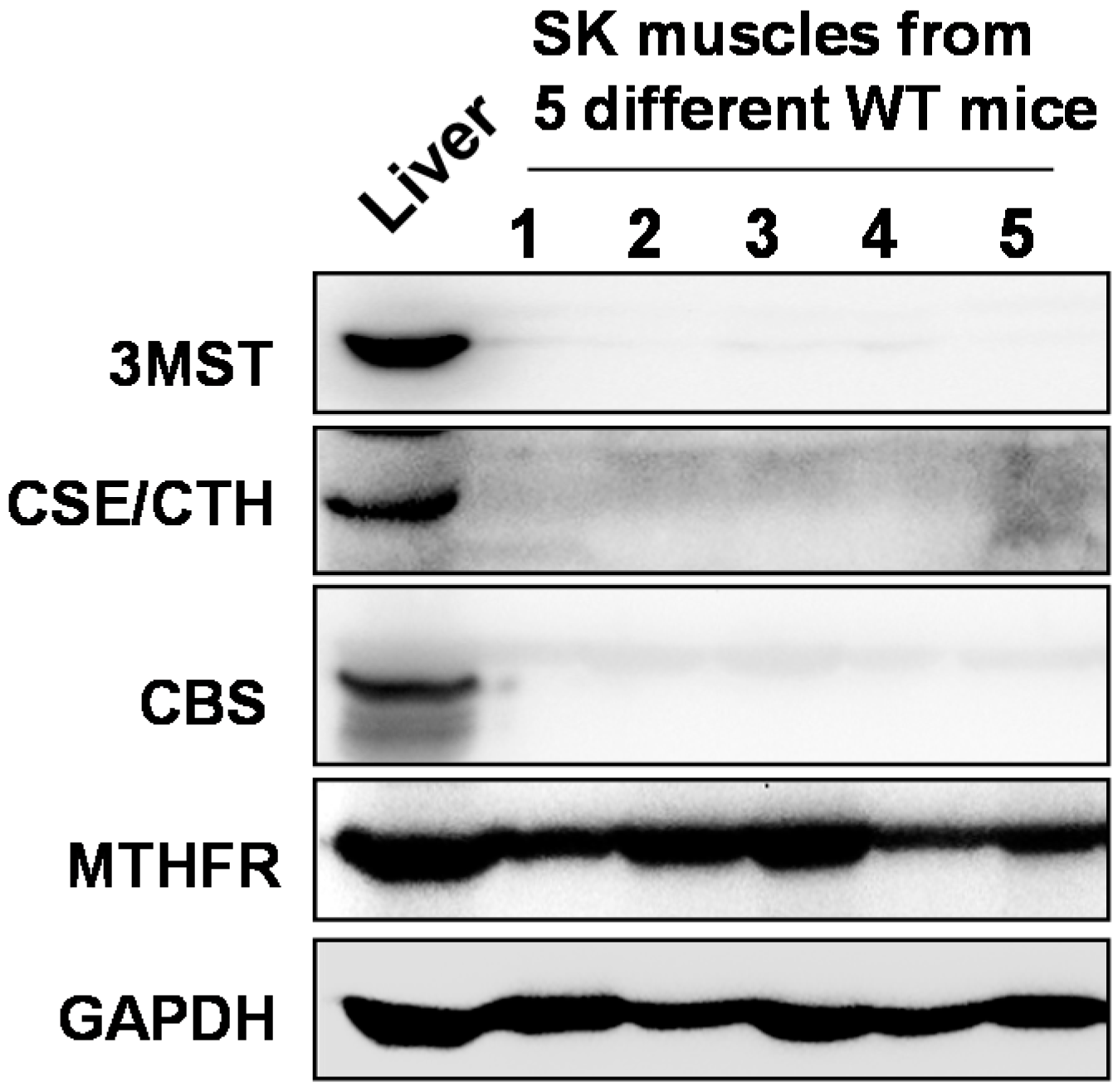
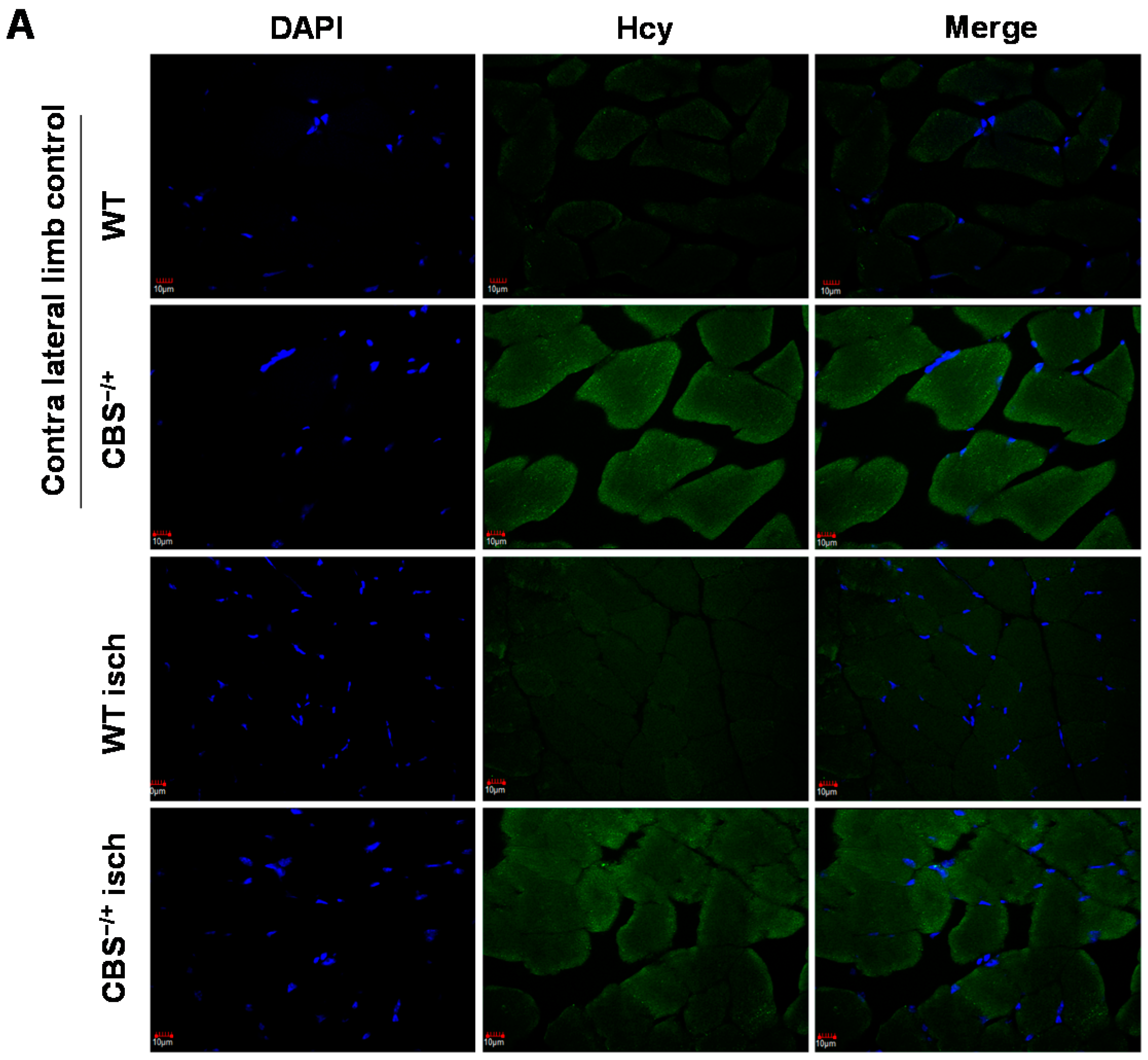
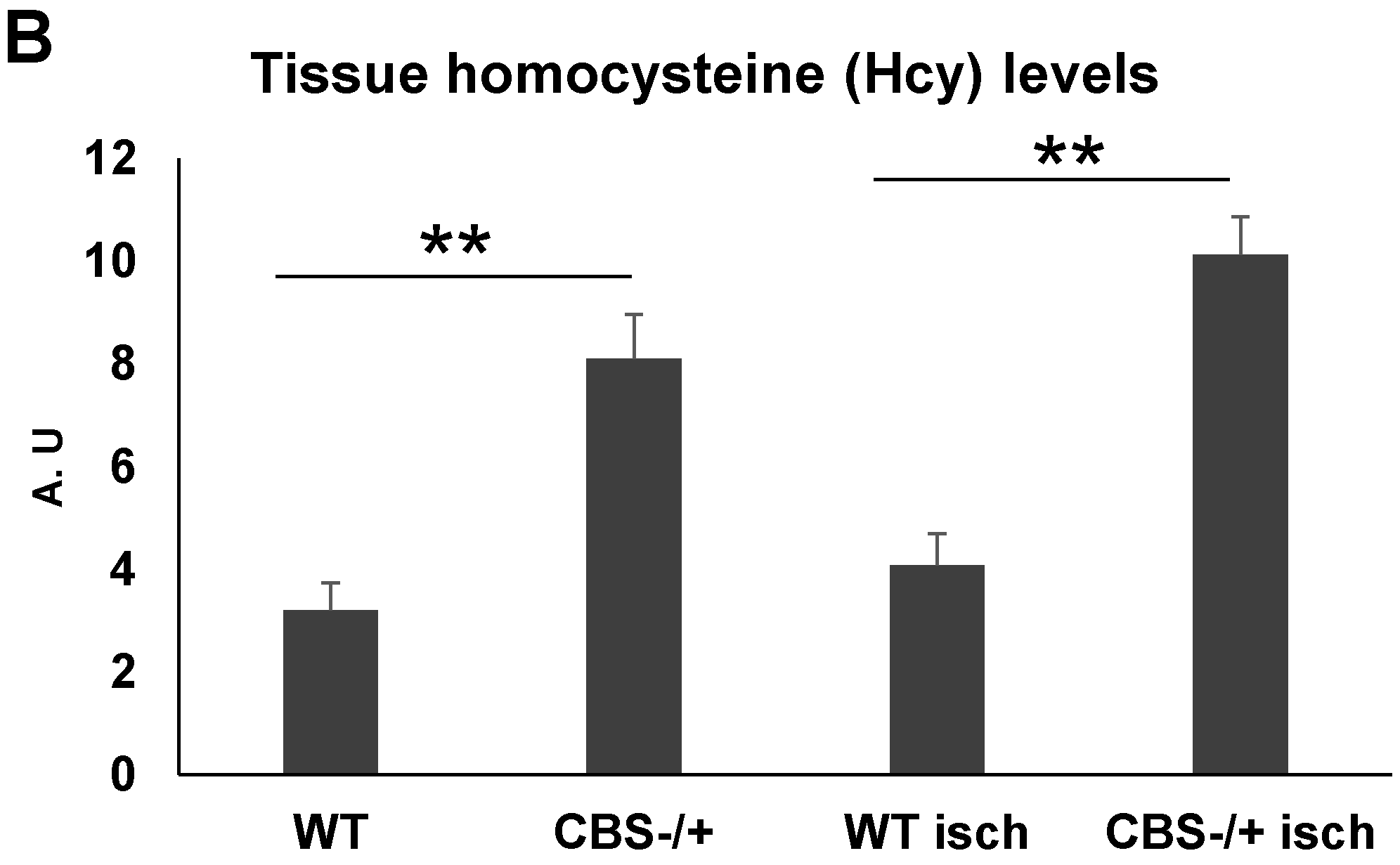
2.1. Skeletal Muscles Lack Hcy Trans-Sulfuration Enzymes

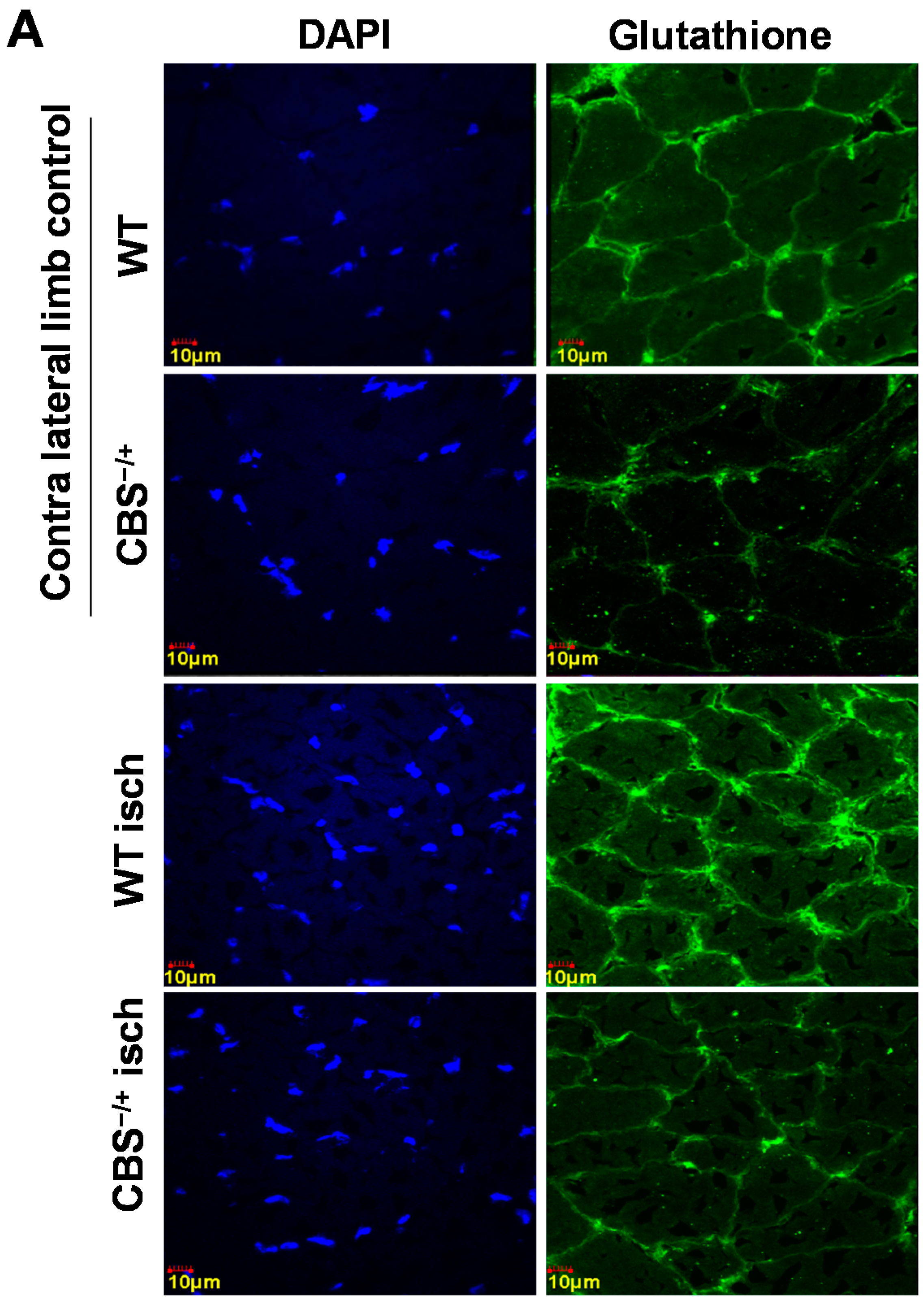
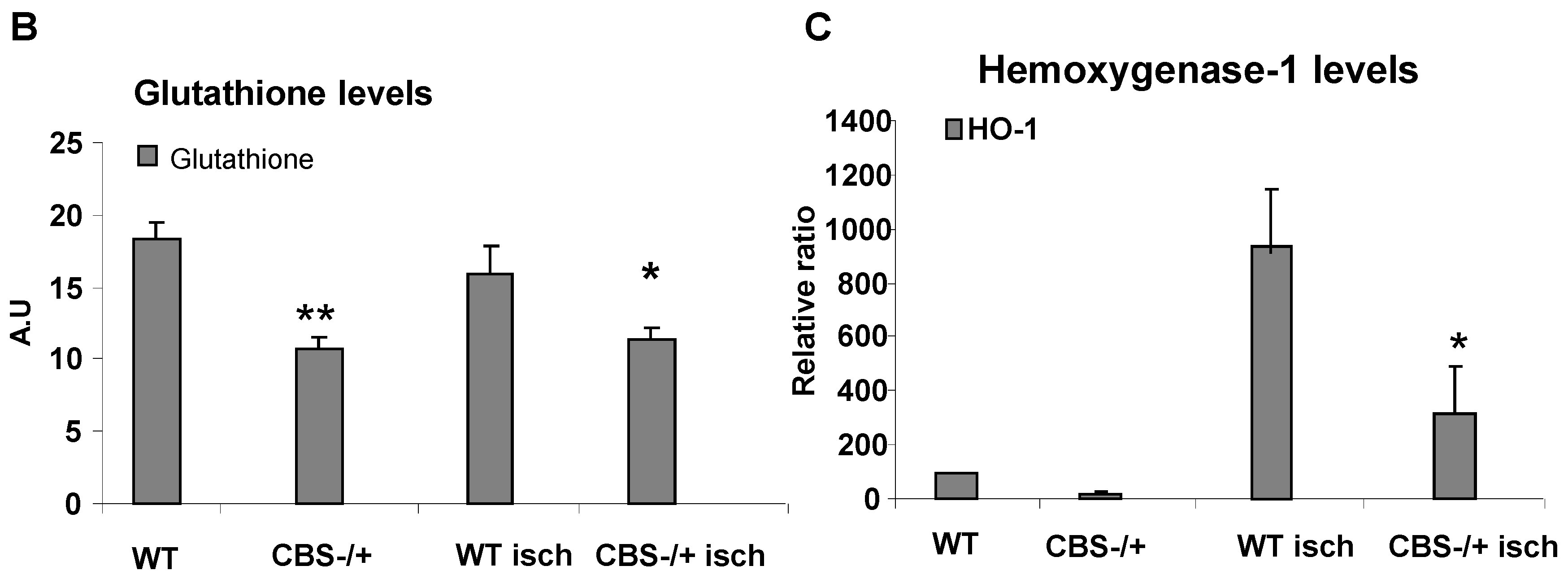
2.2. Attenuated Skeletal Muscle Anti-Oxidant Capacity during HHcy
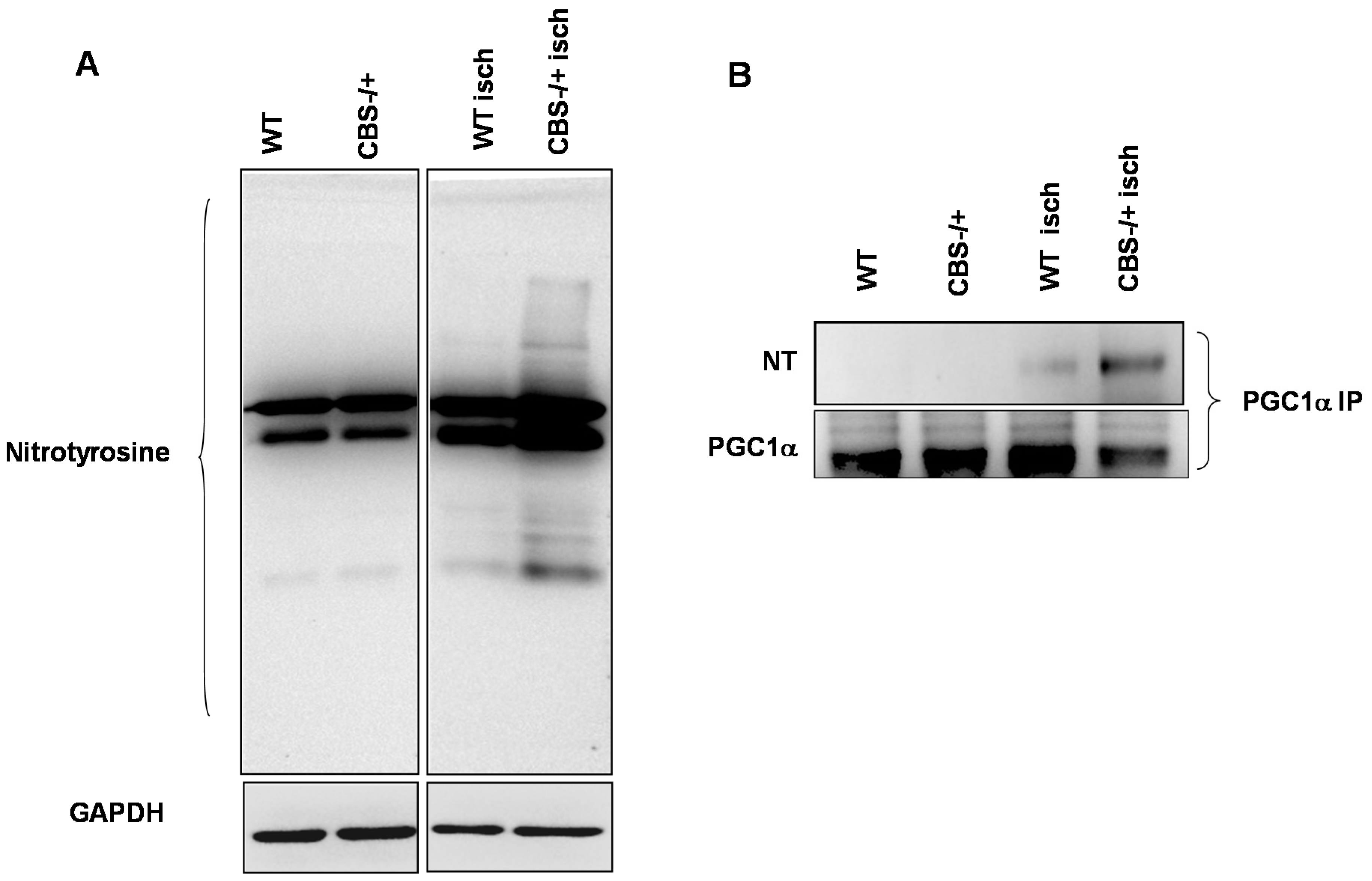
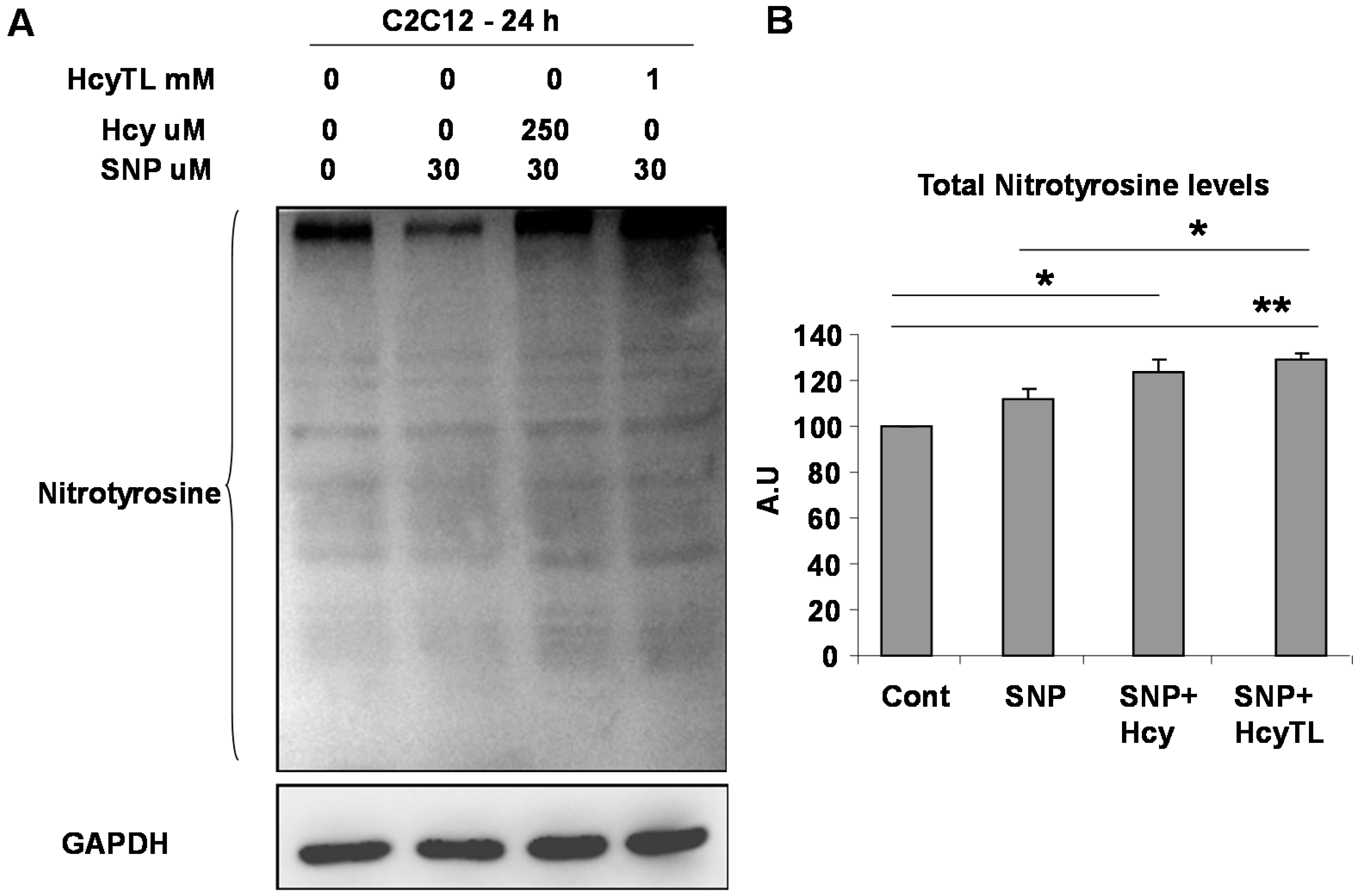
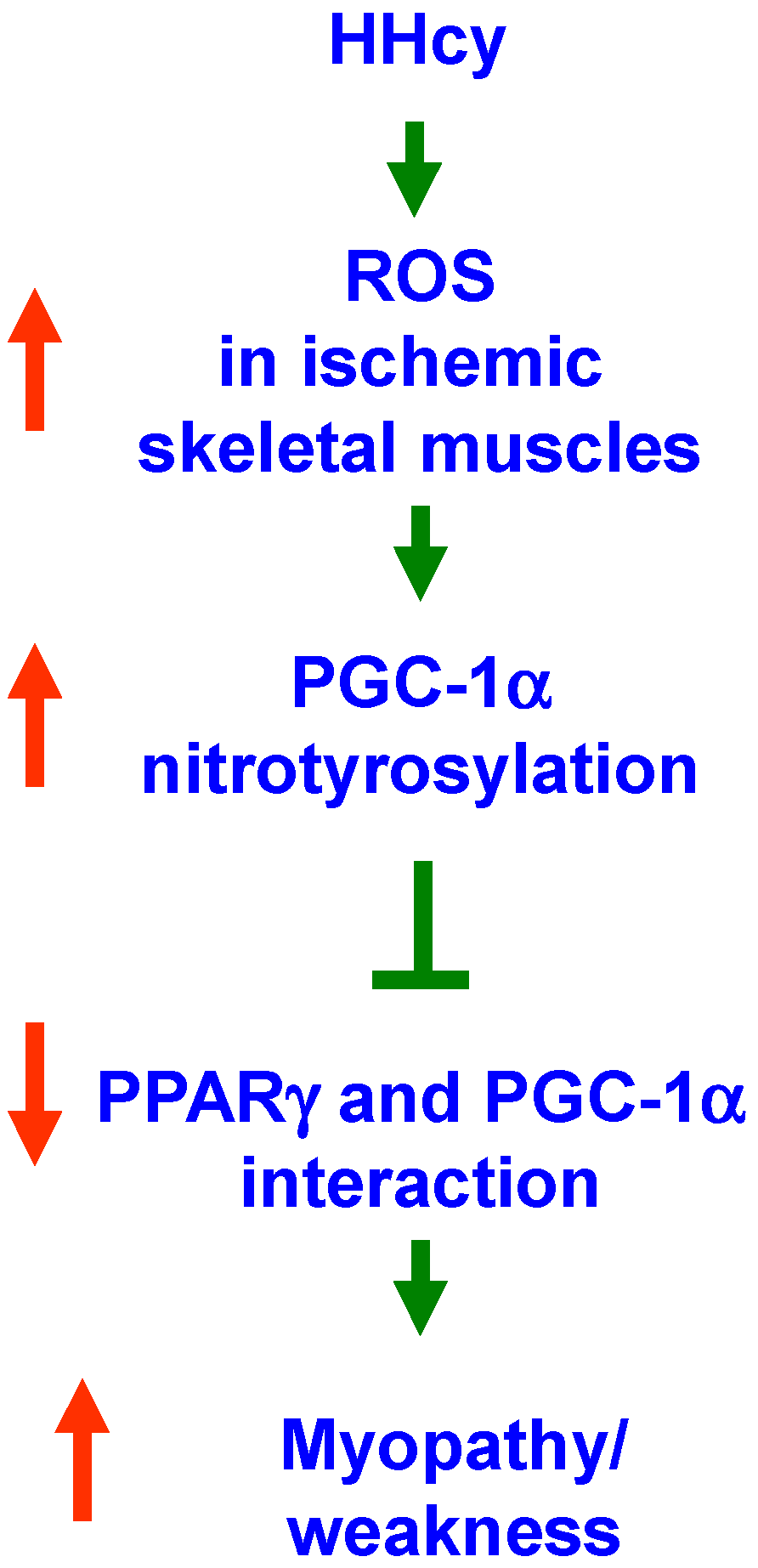
2.3. Enhanced Protein Nitrotyrosylation in Ischemic Skeletal Muscles during HHcy





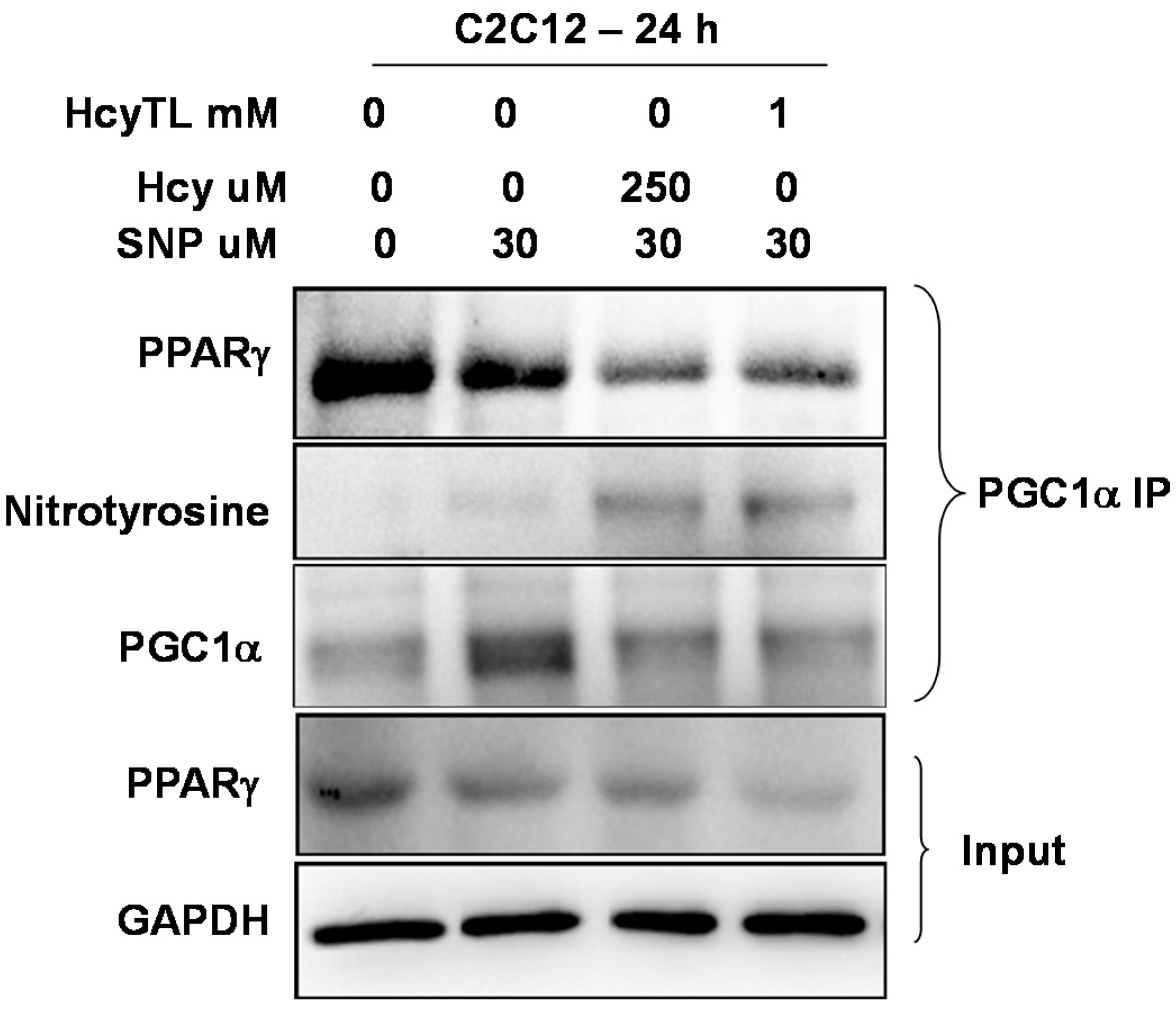
2.4. Inhibition of PGC-1α Interaction with PPARγ in the Presence of Hcy and NO Donor


3. Discussion

4. Materials and Methods
4.1. Animal Care and Tissue Collection
4.2. Cell Culture
4.3. Immunoprecipitation
4.4. Real Time PCR
4.5. Western Blotting
4.6. Antibodies
4.7. Confocal Imaging
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Abbreviation
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1 |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| HHcy | hyperhomocysteinemia |
| Hcy | homocysteine |
| CBS | cystathionine beta-synthase |
| CSE | cystathionine γ-lyase |
| 3MST | 3-mercaptopyruvate sulfurtransferase |
| MTHFR | methylenetetrahydrofolate reductase |
| ROS | Reactive oxygen species |
| HO-1 | heme oxygenase-1 |
| NO | nitric oxide |
| H2S | hydrogen sulfide |
| SNP | Sodium nitroprusside |
Conflicts of Interest
References
- Austin, R.C.; Lentz, S.R.; Werstuck, G.H. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004, 11, S56–S64. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Finkelstein, J.D.; Irreverre, F.; Laster, L. Homocystinuria: An enzymatic defect. Science 1964, 143, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.B.; Carson, N.A.; Neill, D.W. Pathological findings in homocystinuria. J. Clin. Pathol. 1964, 17, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.S.; Carson, N.A. Homocystinuria. The evolution of skeletal changes in relation to treatment. Ann. Radiol. 1978, 21, 95–104. [Google Scholar] [PubMed]
- Tamburrini, O.; Bartolomeo-de Iuri, A.; Andria, G.; Strisciuglio, P.; del Giudice, E.; Palescandolo, P.; Sartorio, R. Bone changes in homocystinuria in childhood. Radiol. Med. 1984, 70, 937–942. [Google Scholar] [PubMed]
- Brosnan, J.T.; Jacobs, R.L.; Stead, L.M.; Brosnan, M.E. Methylation demand: A key determinant of homocysteine metabolism. Acta Biochim. Pol. 2004, 51, 405–413. [Google Scholar] [PubMed]
- Robert, K.; Maurin, N.; Vayssettes, C.; Siauve, N.; Janel, N. Cystathionine beta synthase deficiency affects mouse endochondral ossification. Anat. Rec. A Discov. Mol. Cell. Evolut. Biol. 2005, 282, 1–7. [Google Scholar]
- Holstein, J.H.; Herrmann, M.; Splett, C.; Herrmann, W.; Garcia, P.; Histing, T.; Klein, M.; Kurz, K.; Siebel, T.; Pohlemann, T.; et al. High bone concentrations of homocysteine are associated with altered bone morphology in humans. Br. J. Nutr. 2011, 106, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Kalra, B.R.; Ghose, S.; Sood, N.N. Homocystinuria with bilateral absolute glaucoma. Indian J. Ophthalmol. 1985, 33, 195–197. [Google Scholar] [PubMed]
- Iacobazzi, V.; Infantino, V.; Castegna, A.; Andria, G. Hyperhomocysteinemia: Related genetic diseases and congenital defects, abnormal DNA methylation and newborn screening issues. Mol. Genet. Metab. 2014, 113, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Veeranki, S.; Tyagi, S.C. Defective homocysteine metabolism: Potential implications for skeletal muscle malfunction. Int. J. Mol. Sci. 2013, 14, 15074–15091. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, N.; Pushpakumar, S.B.; Givvimani, S.; Kundu, S.; Metreveli, N.; James, D.; Bratcher, A.P.; Tyagi, S.C. Epigenetic regulation of aortic remodeling in hyperhomocysteinemia. FASEB J. 2014, 28, 3411–3422. [Google Scholar] [CrossRef] [PubMed]
- Signorello, M.; Viviani, G.; Armani, U.; Cerone, R.; Minniti, G.; Piana, A.; Leoncini, G. Homocysteine, reactive oxygen species and nitric oxide in type 2 diabetes mellitus. Thromb. Res. 2007, 120, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Neuman, J.C.; Albright, K.A.; Schalinske, K.L. Exercise prevents hyperhomocysteinemia in a dietary folate-restricted mouse model. Nutr. Res. 2013, 33, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Van Guldener, C. Why is homocysteine elevated in renal failure and what can be expected from homocysteine-lowering? Nephrol. Dial. Transpl. 2006, 21, 1161–1166. [Google Scholar] [CrossRef]
- Sen, U.; Sathnur, P.B.; Kundu, S.; Givvimani, S.; Coley, D.M.; Mishra, P.K.; Qipshidze, N.; Tyagi, N.; Metreveli, N.; Tyagi, S.C. Increased endogenous H2S generation by cbs, cse, and 3mst gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am. J. Physiol. Cell Physiol. 2012, 303, C41–C51. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Edwards, J.G.; Kaminski, P.M.; Wolin, M.S.; Kaley, G.; Koller, A. Increased superoxide production in coronary arteries in hyperhomocysteinemia—Role of tumor necrosis factor-alpha, nad(p)h oxidase, and inducible nitric oxide synthase. Arterioscler. Thromb. Vasc. 2003, 23, 418–424. [Google Scholar] [CrossRef]
- Faraci, F.M. Hyperhomocysteinemia—A million ways to lose control. Arterioscler. Thromb. Vasc. 2003, 23, 371–373. [Google Scholar] [CrossRef]
- Veeranki, S.; Givvimani, S.; Pushpakumar, S.; Tyagi, S.C. Hyperhomocysteinemia attenuates angiogenesis through reduction of hif-1alpha and pgc-1alpha levels in muscle fibers during hindlimb ischemia. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1116–H1127. [Google Scholar] [CrossRef] [PubMed]
- Chintalgattu, V.; Harris, G.S.; Akula, S.A.; Katwa, L.C. Ppar-gamma agonists induce the expression of vegf and its receptors in cultured cardiac myofibroblasts. Cardiovasc. Res. 2007, 74, 140–150. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Ruas, J.; Rao, R.; Kleiner, S.; Wu, J.; Spiegelman, B. A novel pgc-1a isoform induced by resistance training regulates skeletal muscle hypertrophy. FASEB J. 2013, 27, I8. [Google Scholar]
- Handschin, C.; Choi, C.S.; Chin, S.; Kim, S.; Kawamori, D.; Kurpad, A.J.; Neubauer, N.; Hu, J.; Mootha, V.K.; Kim, Y.B.; et al. Abnormal glucose homeostasis in skeletal muscle-specific pgc-1 alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J. Clin. Investig. 2007, 117, 3463–3474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Lin, J.D.; Handschin, C.; Yang, W.L.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. Pgc-1 alpha a protects skeletal muscle from atrophy by suppressing fox03 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rytinki, M.M.; Palvimo, J.J. Sumoylation attenuates the function of pgc-1alpha. J. Biol. Chem. 2009, 284, 26184–26193. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; Pierre, J.; Spiegelman, B.M. Amp-activated protein kinase (ampk) action in skeletal muscle via direct phosphorylation of pgc-1 alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.H.; Monks, B.; Ge, Q.Y.; Birnbaum, M.J. Akt/pkb regulates hepatic metabolism by directly inhibiting pgc-1 alpha transcription coactivator. Nature 2007, 447, U1012–U1018. [Google Scholar] [CrossRef]
- Lee, M.H.; Jang, M.H.; Kim, E.K.; Han, S.W.; Cho, S.Y.; Kim, C.J. Nitric oxide induces apoptosis in mouse c2c12 myoblast cells. J. Pharmacol. Sci. 2005, 97, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Kolling, J.; Scherer, E.B.; Siebert, C.; Marques, E.P.; dos Santos, T.M.; Wyse, A.T. Creatine prevents the imbalance of redox homeostasis caused by homocysteine in skeletal muscle of rats. Gene 2014, 545, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.L.; Zhang, C.Y.; Jin, H.F.; Tang, C.S.; Du, J.B. Hydrogen sulfide regulates lung tissue-oxidized glutathione and total antioxidant capacity in hypoxic pulmonary hypertensive rats. Acta Pharmacol. Sin. 2008, 29, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Tyagi, N.; Sen, U.; Tyagi, S.C. Matrix imbalance by inducing expression of metalloproteinase and oxidative stress in cochlea of hyperhomocysteinemic mice. Mol. Cell. Biochem. 2009, 332, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.J.; Liu, C.A.; Huang, B.; Tseng, A.H.; Wang, D.L. Shear-induced endothelial mechanotransduction: The interplay between reactive oxygen species (ros) and nitric oxide (no) and the pathophysiological implications. J. Biomed. Sci. 2014, 21, 3. [Google Scholar] [CrossRef] [PubMed]
- Salom, M.G.; Arregui, B.; Carbonell, L.F.; Ruiz, F.; Gonzalez- Mora, J.L.; Fenoy, F.J. Renal ischemia induces an increase in nitric oxide levels from tissue stores. Am. J. Physiol. Reg. I 2005, 289, R1459–R1466. [Google Scholar]
- Matsunaga, T.; Warltier, D.C.; Weihrauch, D.W.; Moniz, M.; Tessmer, J.; Chilian, W.M. Ischemia-induced coronary collateral growth is dependent on vascular endothelial growth factor and nitric oxide. Circulation 2000, 102, 3098–3103. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Osada, J.; Aratani, Y.; Kluckman, K.; Reddick, R.; Malinow, M.R.; Maeda, N. Mice deficient in cystathionine beta-synthase—Animal-models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. USA 1995, 92, 1585–1589. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veeranki, S.; Tyagi, S.C. Mechanisms of Hyperhomocysteinemia Induced Skeletal Muscle Myopathy after Ischemia in the CBS−/+ Mouse Model. Int. J. Mol. Sci. 2015, 16, 1252-1265. https://doi.org/10.3390/ijms16011252
Veeranki S, Tyagi SC. Mechanisms of Hyperhomocysteinemia Induced Skeletal Muscle Myopathy after Ischemia in the CBS−/+ Mouse Model. International Journal of Molecular Sciences. 2015; 16(1):1252-1265. https://doi.org/10.3390/ijms16011252
Chicago/Turabian StyleVeeranki, Sudhakar, and Suresh C. Tyagi. 2015. "Mechanisms of Hyperhomocysteinemia Induced Skeletal Muscle Myopathy after Ischemia in the CBS−/+ Mouse Model" International Journal of Molecular Sciences 16, no. 1: 1252-1265. https://doi.org/10.3390/ijms16011252