
Profiling of Ubiquitination Pathway Genes in Peripheral Cells from Patients with Frontotemporal Dementia due to C9ORF72 and GRN Mutations


Abstract
:1. Introduction
2. Results

3. Discussion
4. Materials and Methods
4.1. Patients and Controls

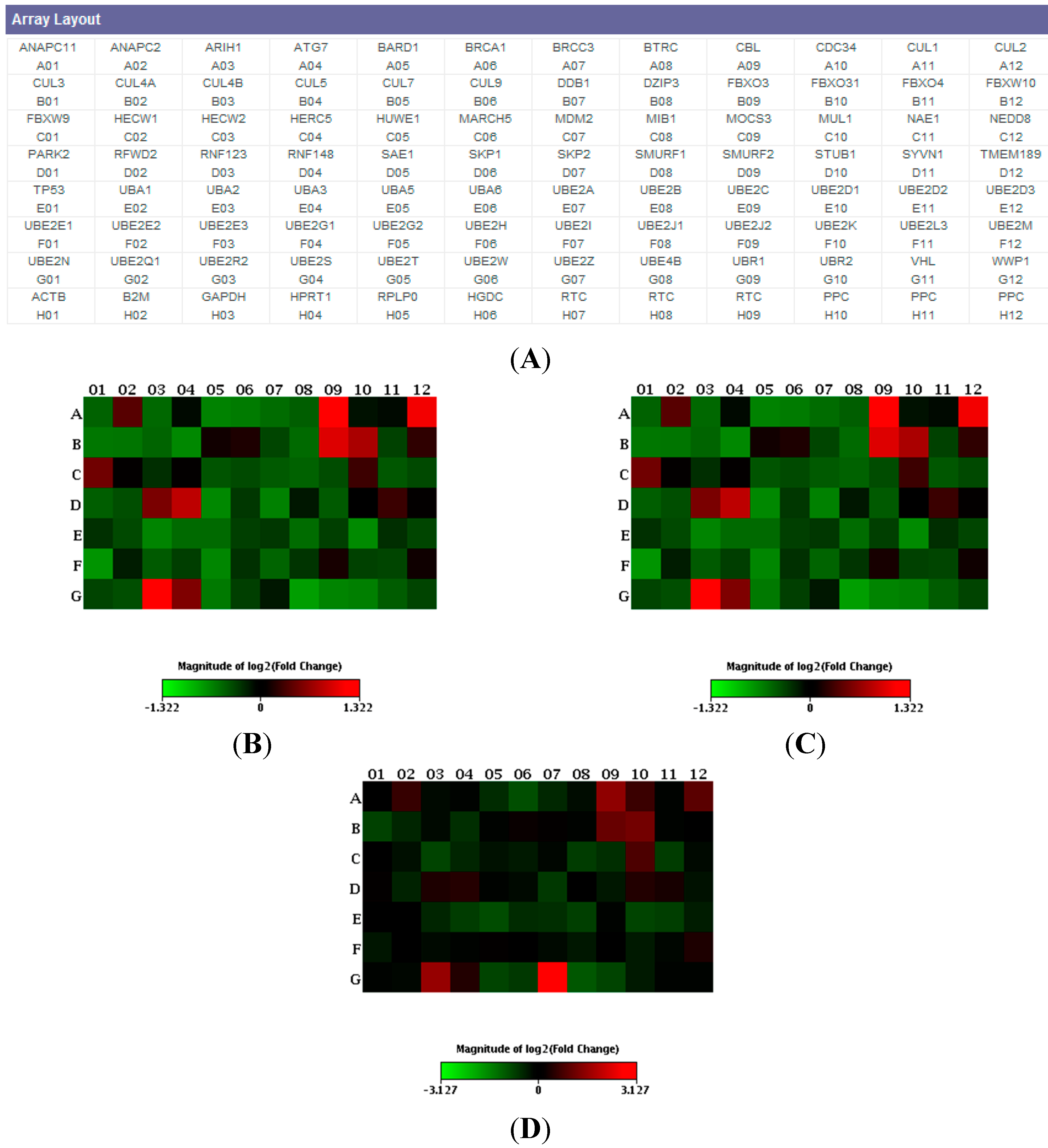{kind=link}
| Characteristics | Sporadic | C9ORF72 | GRN | Controls |
|---|---|---|---|---|
| Number of patients | 6 | 6 | 6 | 6 |
| Gender (M:F) | 4:2 | 5:1 | 4:2 | 3:3 |
| Mean age (years ± SEM) | 66.3 ± 8.6 | 63.9 ± 8.1 | 62 ± 9.4 | 65 ± 7.3 |
| Mean Age at onset (years ± SEM) | 64.5 ± 0.44 | 61 ± 0.36 | 59 ± 0.98 | – |
4.2. Screening of GRN and C9ORF72 Mutations
4.3. Total mRNA Isolation from Peripheral Blood Mononuclear Cells (PBMC)
4.4. Screening of Ubiquitination Pathways by PCR Array
4.5. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seltman, R.E.; Matthews, B.R. Frontotemporal lobar degeneration: Epidemiology, pathology, diagnosis and management. CNS Drugs 2012, 26, 841–870. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010, 119, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Munoz, D.G.; Kusaka, H.; Yokota, O.; Ishihara, K.; Roeber, S.; Kretzschmar, H.A.; Cairns, N.J.; Neumann, M. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 2011, 121, 207–218. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Fenoglio, C.; Serpente, M.; Villa, C.; Bonsi, R.; Arighi, A.; Fumagalli, G.G.; del Bo, R.; Bruni, A.C.; Anfossi, M.; et al. Autosomal dominant frontotemporal lobar degeneration due to the C9ORF72 hexanucleotide repeat expansion: Late-onset psychotic clinical presentation. Biol. Psychiatry 2013, 74, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, J.T.; Sun, F.R.; Ou, J.R.; Qu, S.B.; Tan, L. The clinical and pathological phenotypes of frontotemporal dementia with C9ORF72 mutations. J. Neurol. Sci. 2013, 335, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A.; Hortobágyi, T.; Shaw, C.E. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011, 122, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; Tabrizi, S.J. The ubiquitin-proteasome system in neurodegeneration. Antioxid. Redox. Signal. 2014, 21, 2302–2321. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, U.; Takahashi, S.; Ishizu, A.; Miyatake, Y.; Gohda, A.; Suzuki, S.; Ono, A.; Ohara, J.; Baba, T.; Murata, S.; et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 2012, 180, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Morantte, I.; Liu, Z.; Douglas, P.M.; Merkwirth, C.; Rodrigues, A.P.; Manning, G.; Dillin, A. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 2012, 489, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A. Ubiquitin: Roles in protein modification and breakdown. Cell 1983, 34, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [PubMed]
- Hochstrasser, M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol. 1995, 7, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Rubino, E.; Rainero, I.; Chiò, A.; Rogaeva, E.; Galimberti, D.; Fenoglio, P.; Grinberg, Y.; Isaia, G.; Calvo, A.; Gentile, S.; et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012, 79, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Le Ber, I.; Camuzat, A.; Guerreiro, R.; Bouya-Ahmed, K.; Bras, J.; Nicolas, G.; Gabelle, A.; Didic, M.; de Septenville, A.; Millecamps, S.; et al. SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 2013, 70, 1403–1410. [Google Scholar] [PubMed]
- Van der Zee, J.; van Langenhove, T.; Kovacs, G.G.; Dillen, L.; Deschamps, W.; Engelborghs, S.; Matěj, R.; Vandenbulcke, M.; Sieben, A.; Dermaut, B.; et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 2014, 128, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, G.P.; Alves, C.J.; Chadi, G. Early gene expression changes in spinal cord from SOD1(G93A) Amyotrophic Lateral Sclerosis animal model. Front. Cell. Neurosci. 2013, 7, 216. [Google Scholar] [PubMed]
- Hanus, C.; Schuman, E.M. Proteostasis in complex dendrites. Nat. Rev. Neurosci. 2013, 14, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Figueiredo-Pereira, M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: Therapeutic implications. Apoptosis 2010, 15, 1292–1311. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Chen, J.; Hu, B.; Zou, H.; Li, A.; Guo, A.; Jiang, J. Down-regulation of UBE2Q1 is associated with neuronal apoptosis in rat brain cortex following traumatic brain injury. J. Neurosci. Res. 2014, 92, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhao, F.; Zheng, M.; Fei, X.; Chen, X.; Huang, S.; Xie, Y.; Mao, Y. Cloning and characterization of a gene encoding the human putative ubiquitin conjugating enzyme E2Z (UBE2Z). Mol. Biol. Rep. 2007, 34, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.Y.; Sung, C.C.; Brito, I.L.; Sheng, M. Degradation of postsynaptic scaffold GKAP and regulation of dendritic spine morphology by the TRIM3 ubiquitin ligase in rat hippocampal neurons. PLoS One 2010, 5, e9842. [Google Scholar] [CrossRef] [PubMed]
- Hans, F.; Fiesel, F.C.; Strong, J.C.; Jäckel, S.; Rasse, T.M.; Geisler, S.; Springer, W.; Schulz, J.B.; Voigt, A.; Kahle, P.J. UBE2E ubiquitin-conjugating enzymes and ubiquitin isopeptidase Y regulate TDP-43 protein ubiquitination. J. Biol. Chem. 2014, 289, 19164–19179. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.K.; Hung, K.W.; Fu, W.Y.; Shen, C.; Chen, Y.; Xia, J.; Lai, K.O.; Ip, N.Y. APC (Cdh1) mediates EphA4-dependent down-regulation of AMPA receptors in homeostatic plasticity. Nat. Neurosci. 2011, 14, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration. A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Albert, M.S.; Grossman, M.; Miller, B.; Dickson, D.; Trojanowski, J.Q. Clinical and pathological diagnosis of frontotemporal dementia: Report of the work group on frontotemporal dementia and Pick’s disease. Arch. Neurol. 2001, 58, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef] [PubMed]
- Pietroboni, A.M.; Fumagalli, G.G.; Ghezzi, L.; Fenoglio, C.; Cortini, F.; Serpente, M.; Cantoni, C.; Rotondo, E.; Corti, P.; Carecchio, M.; et al. Phenotypic heterogeneity of the GRN Asp22fs mutation in a large Italian kindred. J. Alzheimers Dis. 2011, 24, 253–259. [Google Scholar] [PubMed]
- Dobson-Stone, C.; Hallupp, M.; Bartley, L.; Shepherd, C.E.; Halliday, G.M.; Schofield, P.R.; Hodges, J.R.; Kwok, J.B. C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology 2012, 79, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Serpente, M.; Fenoglio, C.; Villa, C.; Cortini, F.; Cantoni, C.; Ridolfi, E.; Clerici, F.; Marcone, A.; Benussi, L.; Ghidoni, R.; et al. Role of OLR1 and its regulating hsa-miR369–3p in Alzheimer’s disease: Genetics and expression analysis. J. Alzheimers Dis. 2011, 26, 787–793. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serpente, M.; Fenoglio, C.; Cioffi, S.M.G.; Bonsi, R.; Arighi, A.; Fumagalli, G.G.; Ghezzi, L.; Scarpini, E.; Galimberti, D. Profiling of Ubiquitination Pathway Genes in Peripheral Cells from Patients with Frontotemporal Dementia due to C9ORF72 and GRN Mutations. Int. J. Mol. Sci. 2015, 16, 1385-1394. https://doi.org/10.3390/ijms16011385
Serpente M, Fenoglio C, Cioffi SMG, Bonsi R, Arighi A, Fumagalli GG, Ghezzi L, Scarpini E, Galimberti D. Profiling of Ubiquitination Pathway Genes in Peripheral Cells from Patients with Frontotemporal Dementia due to C9ORF72 and GRN Mutations. International Journal of Molecular Sciences. 2015; 16(1):1385-1394. https://doi.org/10.3390/ijms16011385
Chicago/Turabian StyleSerpente, Maria, Chiara Fenoglio, Sara M. G. Cioffi, Rossana Bonsi, Andrea Arighi, Giorgio G. Fumagalli, Laura Ghezzi, Elio Scarpini, and Daniela Galimberti. 2015. "Profiling of Ubiquitination Pathway Genes in Peripheral Cells from Patients with Frontotemporal Dementia due to C9ORF72 and GRN Mutations" International Journal of Molecular Sciences 16, no. 1: 1385-1394. https://doi.org/10.3390/ijms16011385
APA StyleSerpente, M., Fenoglio, C., Cioffi, S. M. G., Bonsi, R., Arighi, A., Fumagalli, G. G., Ghezzi, L., Scarpini, E., & Galimberti, D. (2015). Profiling of Ubiquitination Pathway Genes in Peripheral Cells from Patients with Frontotemporal Dementia due to C9ORF72 and GRN Mutations. International Journal of Molecular Sciences, 16(1), 1385-1394. https://doi.org/10.3390/ijms16011385