
Comparative Proteomic Analysis of Gossypium thurberi in Response to Verticillium dahliae Inoculation


Abstract
:1. Introduction
2. Results and Discussion
2.1. Colonization Identification of V. dahliae in Seedling Roots after Inoculation

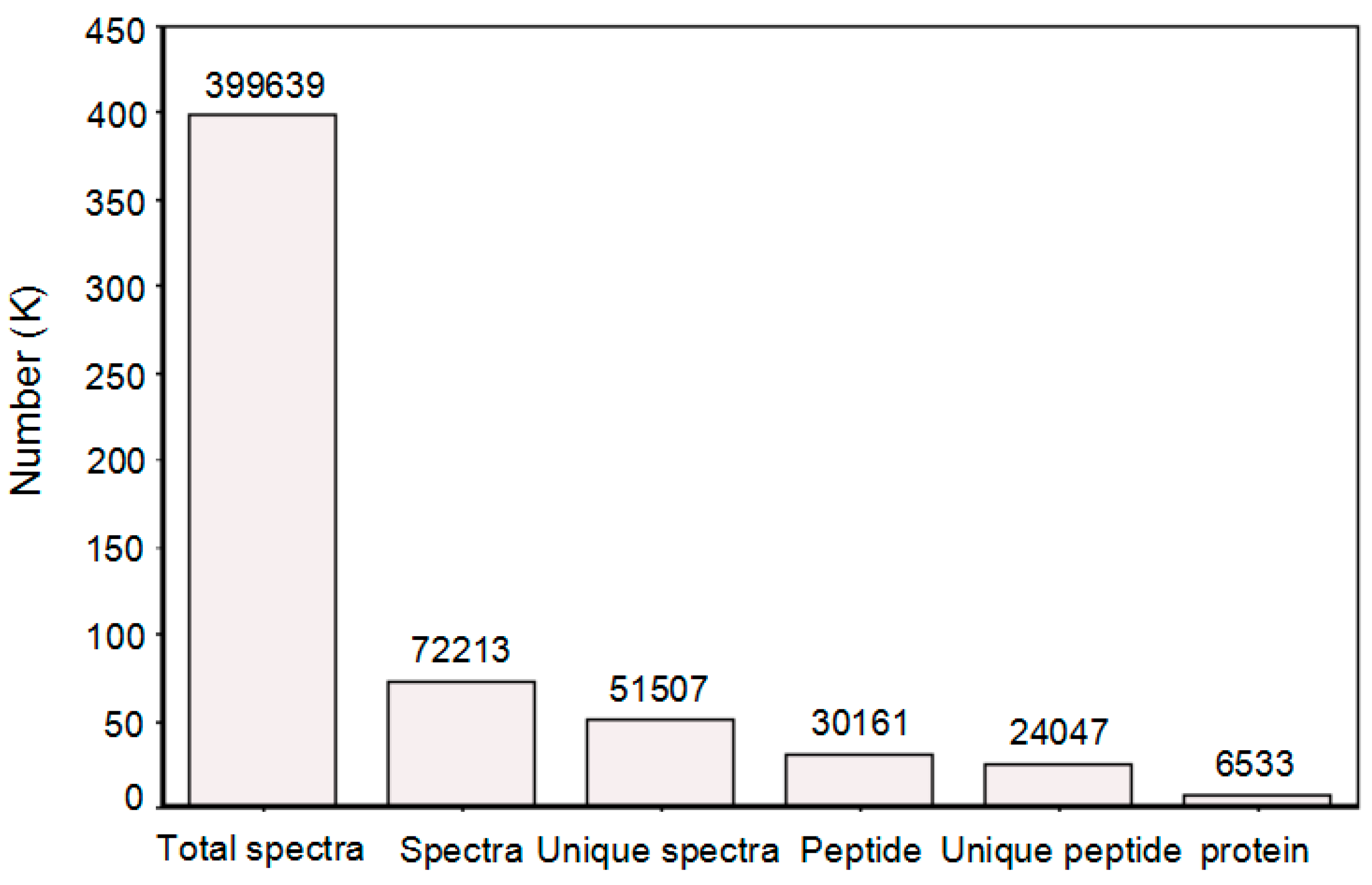
2.2. Proteins Identified by I-TRAQ and Their Functional Annotation


2.3. Identification and Enrichment Analysis of Differentially Expressed Proteins (DEPs)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Enriched KEGG Pathways | DEPs | p-Value | |
|---|---|---|---|---|
| Up (304) | Down (204) | |||
| 1 | Metabolic pathways (ko01100) | 104 (34.21%) | 79 (38.73%) | 1 |
| 2 | Biosynthesis of secondary metabolites (ko01110) | 69 (22.7%) | 52 (25.49%) | 1 |
| 3 | Ribosome (ko03010) | 32 (10.53%) | 20 (9.8%) | 1 |
| 4 | Protein processing in endoplasmic reticulum (ko04141) | 24 (7.89%) | 2 (0.98%) | 1 |
| 5 | Starch and sucrose metabolism (ko00500) | 21 (6.91%) | 5 (2.45%) | 1 |
| 6 | Protein export (ko03060) | 1 (0.33%) | 3 (1.47%) | 1 |
| 7 | Phenylpropanoid biosynthesis (ko00940) | 16 (5.26%) | 0 | 1 |
| 8 | Spliceosome (ko03040) | 13 (4.28%) | 13 (6.37%) | 1 |
| 9 | Flavonoid biosynthesis (ko00941) | 12 (3.95%) | 9 (4.41%) | 1 |
| 10 | Phenylalanine metabolism (ko00360) | 11 (3.62%) | 10 (4.9%) | 1 |
| 11 | Plant-pathogen interaction (ko04626) | 10 (3.29%) | 2 (0.98%) | 1 |
| 12 | Glycolysis/Gluconeogenesis (ko00010) | 9 (2.96%) | 4 (1.96%) | 1 |
| 13 | Phagosome (ko04145) | 8 (2.63%) | 2 (0.98%) | 1 |
| 14 | Endocytosis (ko04144) | 7 (2.3%) | 2 (0.98%) | 1 |
| 15 | Amino sugar and nucleotide sugar metabolism (ko00520) | 7 (2.3%) | 7 (3.43%) | 1 |
| 16 | Oxidative phosphorylation (ko00190) | 7 (2.3%) | 14 (6.86%) | 1 |
| 17 | RNA transport (ko03013) | 7 (2.3%) | 5 (2.45%) | 1 |
| 18 | Fatty acid metabolism (ko00071) | 7 (2.3%) | 2 (0.98%) | 1 |
| 19 | Ascorbate and aldarate metabolism (ko00053) | 7 (2.3%) | 6 (2.94%) | 1 |
| 20 | Aminoacyl-tRNA biosynthesis (ko00970) | 7 (2.3%) | 17 (8.33%) | 1 |
| 21 | Carbon fixation in photosynthetic organisms (ko00710) | 7 (2.3%) | 0 | 1 |
| 22 | Glyoxylate and dicarboxylate metabolism (ko00630) | 6 (1.97%) | 0 | 1 |
| 23 | Nitrogen metabolism (ko00910) | 6 (1.97%) | 3 (1.47%) | 1 |
| 24 | Alanine, aspartate and glutamate metabolism (ko00250) | 6 (1.97%) | 1 (0.49%) | 1 |
| 25 | Galactose metabolism (ko00052) | 6 (1.97%) | 4 (1.96%) | 1 |
| 26 | Pyruvate metabolism (ko00620) | 6 (1.97%) | 4 (1.96%) | 1 |
| 27 | Plant hormone signal transduction (ko04075) | 5 (1.64%) | 1 (0.49%) | 1 |
| 28 | Propanoate metabolism (ko00640) | 5 (1.64%) | 4 (1.96%) | 1 |
| 29 | Stilbenoid, diarylheptanoid and gingerol biosynthesis (ko00945) | 5 (1.64%) | 7 (3.43%) | 1 |
| 30 | Pyrimidine metabolism (ko00240) | 5 (1.64%) | 6 (2.94%) | 1 |
| 31 | Glycerophospholipid metabolism (ko00564) | 5 (1.64%) | 1 (0.49%) | 1 |
| 32 | Pentose phosphate pathway (ko00030) | 4 (1.32%) | 1 (0.49%) | 1 |
| 33 | Peroxisome (ko04146) | 4 (1.32%) | 3 (1.47%) | 1 |
| 34 | Limonene and pinene degradation (ko00903) | 4 (1.32%) | 6 (2.94%) | 1 |
| 35 | Terpenoid backbone biosynthesis (ko00900) | 4 (1.32%) | 1 (0.49%) | 1 |
| 36 | Tryptophan metabolism (ko00380) | 4 (1.32%) | 1 (0.49%) | 1 |
| 37 | Cutin, suberine and wax biosynthesis (ko00073) | 4 (1.32%) | 1 (0.49%) | 1 |
| 38 | Proteasome (ko03050) | 4 (1.32%) | 3 (1.47%) | 1 |
| 39 | Pentose and glucuronate interconversions (ko00040) | 4 (1.32%) | 4 (1.96%) | 1 |
| 40 | Tyrosine metabolism (ko00350) | 4 (1.32%) | 0 | 1 |
| 41 | Purine metabolism (ko00230) | 3 (0.99%) | 8 (3.92%) | 1 |
| 42 | Porphyrin and chlorophyll metabolism (ko00860) | 3 (0.99%) | 1 (0.49%) | 1 |
| 43 | Glucosinolate biosynthesis (ko00966) | 3 (0.99%) | 0 | 1 |
| 44 | mRNA surveillance pathway (ko03015) | 3 (0.99%) | 4 (1.96%) | 1 |
| 45 | Flavone and flavonol biosynthesis (ko00944) | 3 (0.99%) | 8 (3.92%) | 1 |
| 46 | Citrate cycle (ko00020) | 3 (0.99%) | 1 (0.49%) | 1 |
| 47 | Glycerolipid metabolism (ko00561) | 3 (0.99%) | 2 (0.98%) | 1 |
| 48 | Phosphatidylinositol signaling system (ko04070) | 3 (0.99%) | 0 | 1 |
| 49 | Ubiquinone and other terpenoid-quinone biosynthesis (ko00130) | 3 (0.99%) | 0 | 1 |
| 50 | Diterpenoid biosynthesis (ko00904) | 3 (0.99%) | 7 (3.43%) | 1 |
| 51 | β-Alanine metabolism (ko00410) | 3 (0.99%) | 3 (1.47%) | 1 |
| 52 | Ubiquitin mediated proteolysis (ko04120) | 3 (0.99%) | 0 | 1 |
| 53 | Glycine, serine and threonine metabolism (ko00260) | 3 (0.99%) | 1 (0.49%) | 1 |
| 54 | Fatty acid biosynthesis (ko00061) | 2 (0.66%) | 1 (0.49%) | 1 |
| 55 | Lysine degradation (ko00310) | 2 (0.66%) | 1 (0.49%) | 1 |
| 56 | Cysteine and methionine metabolism (ko00270) | 2 (0.66%) | 2 (0.98%) | 1 |
| 57 | Other glycan degradation (ko00511) | 2 (0.66%) | 1 (0.49%) | 1 |
| 58 | Monoterpenoid biosynthesis (ko00902) | 2 (0.66%) | 0 | 1 |
| 59 | Phenylalanine, tyrosine and tryptophan biosynthesis (ko00400) | 2 (0.66%) | 0 | 1 |
| 60 | Sphingolipid metabolism (ko00600) | 2 (0.66%) | 2 (0.98%) | 1 |
| 61 | Arginine and proline metabolism (ko00330) | 2 (0.66%) | 1 (0.49%) | 1 |
| 62 | Nucleotide excision repair (ko03420) | 2 (0.66%) | 1 (0.49%) | 1 |
| 63 | Sulfur metabolism (ko00920) | 2 (0.66%) | 0 | 1 |
| 64 | Glycosphingolipid biosynthesis-ganglio series (ko00604) | 1 (0.33%) | 1 (0.49%) | 1 |
| 65 | ABC transporters (ko02010) | 1 (0.33%) | 1 (0.49%) | 1 |
| 66 | α-Linolenic acid metabolism (ko00592) | 1 (0.33%) | 3 (1.47%) | 1 |
| 67 | Photosynthesis (ko00195) | 1 (0.33%) | 0 | 1 |
| 68 | Tropane, piperidine and pyridine alkaloid biosynthesis (ko00960) | 1 (0.33%) | 2 (0.98%) | 1 |
| 69 | Inositol phosphate metabolism (ko00562) | 1 (0.33%) | 0 | 1 |
| 70 | Anthocyanin biosynthesis (ko00942) | 1 (0.33%) | 0 | 1 |
| 71 | Pantothenate and CoA biosynthesis (ko00770) | 1 (0.33%) | 0 | 1 |
| 72 | Glycosaminoglycan degradation (ko00531) | 1 (0.33%) | 1 (0.49%) | 1 |
| 73 | Taurine and hypotaurine metabolism (ko00430) | 1 (0.33%) | 0 | 1 |
| 74 | Histidine metabolism (ko00340) | 1 (0.33%) | 1 (0.49%) | 1 |
| 75 | Biosynthesis of unsaturated fatty acids (ko01040) | 1 (0.33%) | 0 | 1 |
| 76 | Butanoate metabolism (ko00650) | 1 (0.33%) | 1 (0.49%) | 1 |
| 77 | Benzoxazinoid biosynthesis (ko00402) | 1 (0.33%) | 1 (0.49%) | 1 |
| 78 | Fatty acid elongation (ko00062) | 1 (0.33%) | 0 | 1 |
| 79 | DNA replication (ko03030) | 1 (0.33%) | 1 (0.49%) | 1 |
| 80 | Isoquinoline alkaloid biosynthesis (ko00950) | 1 (0.33%) | 0 | 1 |
| 81 | Glutathione metabolism (ko00480) | 1 (0.33%) | 7 (3.43%) | 1 |
| 82 | Valine, leucine and isoleucine degradation (ko00280) | 1 (0.33%) | 3 (1.47%) | 1 |
| 83 | RNA polymerase (ko03020) | 1 (0.33%) | 4 (1.96%) | 1 |
| 84 | Fructose and mannose metabolism (ko00051) | 1 (0.33%) | 2 (0.98%) | 1 |
| 85 | Cyanoamino acid metabolism (ko00460) | 1 (0.33%) | 0 | 1 |
| 86 | One carbon pool by folate (ko00670) | 1 (0.33%) | 0 | 1 |
| 87 | SNARE interactions in vesicular transport (ko04130) | 1 (0.33%) | 1 (0.49%) | 1 |
| 88 | Regulation of autophagy (ko04140) | 1 (0.33%) | 0 | 1 |
| 89 | RNA degradation (ko03018) | 1 (0.33%) | 2 (0.98%) | 1 |
| 90 | Circadian rhythm-plant (ko04712) | 1 (0.33%) | 0 | 1 |
| 91 | Zeatin biosynthesis (ko00908) | 0 | 3 (1.47%) | 1 |
| 92 | Isoflavonoid biosynthesis (ko00943) | 0 | 3 (1.47%) | 1 |
| 93 | Selenocompound metabolism (ko00450) | 0 | 2 (0.98%) | 1 |
| 94 | Sesquiterpenoid and triterpenoid biosynthesis (ko00909) | 0 | 1 (0.49%) | 1 |
| 95 | Ribosome biogenesis in eukaryotes (ko03008) | 0 | 4 (1.96%) | 1 |
| 96 | Indole alkaloid biosynthesis (ko00901) | 0 | 1 (0.49%) | 1 |
| 97 | Brassinosteroid biosynthesis (ko00905) | 0 | 2 (0.98%) | 1 |
| 98 | Steroid biosynthesis (ko00100) | 0 | 1 (0.49%) | 1 |
| 99 | Riboflavin metabolism (ko00740) | 0 | 1 (0.49%) | 1 |
| 100 | Valine, leucine and isoleucine biosynthesis (ko00290) | 0 | 1 (0.49%) | 1 |
| 101 | Vitamin B6 metabolism (ko00750) | 0 | 2 (0.98%) | 1 |
| 102 | Base excision repair (ko03410) | 0 | 1 (0.49%) | 1 |
2.4. Proteins Related to Cell Wall Organization and Reinforcement

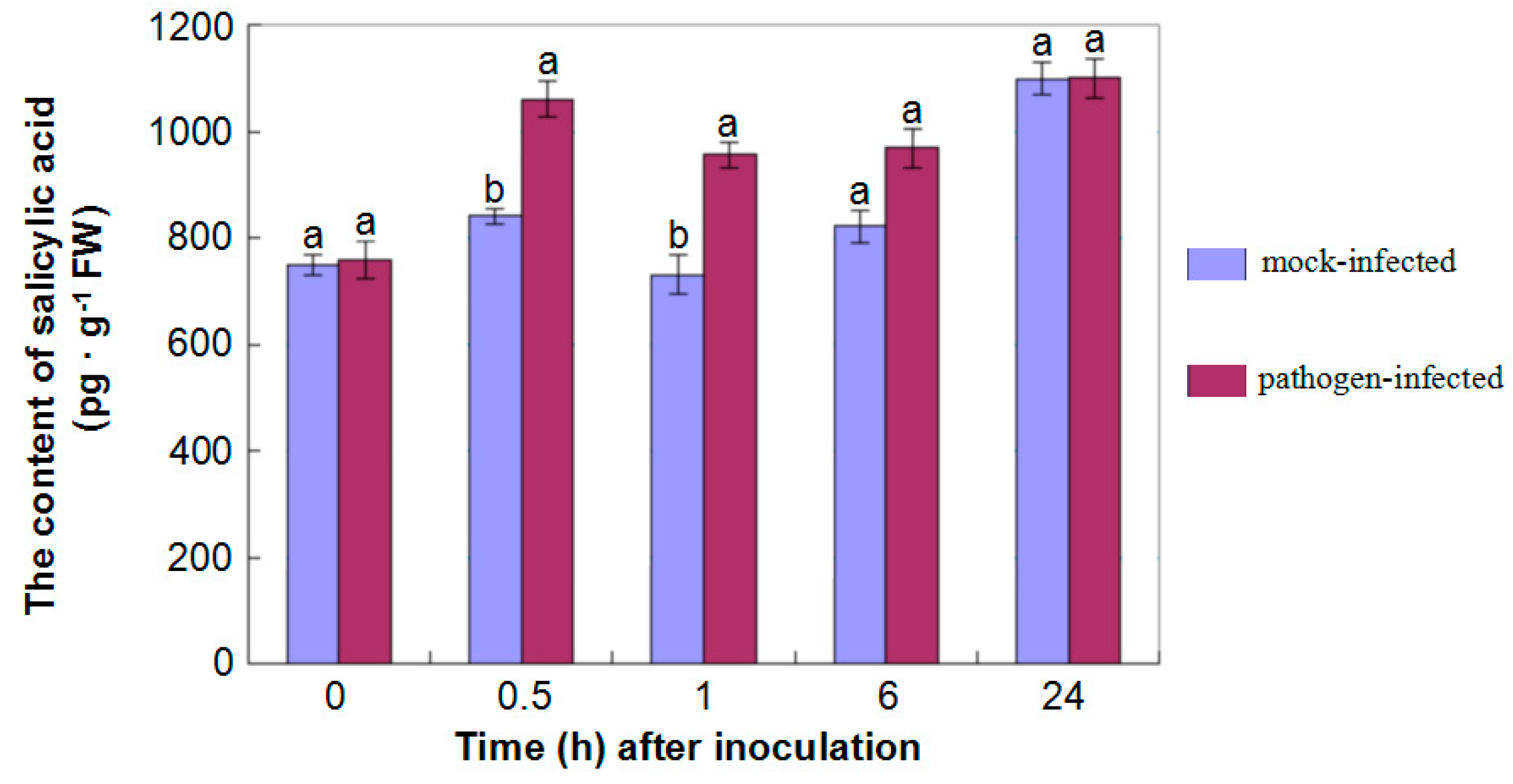
2.5. Products of Metabolism Involved in Response of G. thurberi to V. dahliae
2.6. The Differential Expression of Proteins in Signal Pathways of Phytohormones

2.7. Disease Resistance Response Proteins

3. Experimental Section
3.1. Plant Cultivation and Inoculation
3.2. Protein Extraction and Quantification
3.3. I-TRAQ Labeling and SCX Fractionation
3.4. LC-ESI-MS/MS and Data Analysis
3.5. Bioinformatics Analysis
3.6. Measurement of Enzyme Activity
3.7. Content Quantification of Hormones
3.8. qRT-PCR Analysis
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Song, X.L.; Zhang, T.Z. Identification of quantitative trait loci controlling seed physical and nutrient traits in cotton. Seed Sci. Res. 2007, 17, 243–251. [Google Scholar] [CrossRef]
- Sink, K.; Grey, W.E. A root-injection method to assess Verticillium wilt resistance of peppermint (Mentha × piperita L) and its use in identifying resistant somaclones of cv. Black Mitcham. Euphytica 1999, 106, 223–230. [Google Scholar] [CrossRef]
- Klosterman, S.J.; Atallah, Z.K.; Vallad, G.E.; Subbarao, K.V. Diversity, pathogenicity, and management of Verticillium species. Annu. Rev. Phytopathol. 2009, 47, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Bejarano Alcazar, J.; Blanco Lopez, M.A.; Melero Vara, J.M.; Jimenez Diaz, R.M. The influence of Verticillium wilt epidemics on cotton yield in southern Spain. Plant Pathol. 1997, 46, 168–178. [Google Scholar] [CrossRef]
- Cai, Y.F.; He, X.H.; Mo, J.C.; Sun, Q.; Yang, J.P.; Liu, J.G. Molecular research and genetic engineering of resistance to Verticillium wilt in cotton. Afr. J. Biotechnol. 2009, 8, 7363–7372. [Google Scholar]
- Kamal, M.E. Integrated control of Verticillium wilt of cotton. Plant Dis. 1985, 69, 1025–1032. [Google Scholar]
- Yang, C.; Guo, W.Z.; Li, G.Y.; Gao, F.; Lin, S.S.; Zhang, T.Z. QTLs mapping for Verticillium wilt resistance at seedling and maturity stages in Gossypium barbadense L. Plant Sci. 2007, 174, 290–298. [Google Scholar] [CrossRef]
- Zhao, F.A.; Fang, W.P.; Xie, D.Y.; Zhao, Y.M.; Tang, Z.J.; Li, W.; Nie, L.H.; Lv, S.P. Proteomic identification of differentially expressed proteins in Gossypium thurberi inoculated with cotton Verticillium dahliae. Plant Sci. 2012, 185–186, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Lu, S.; Xu, Y.H.; Wang, J.W.; Chen, X.Y. A cotton cDNA (GaPR-10) encoding a pathogenesis-related 10 protein with in vitro ribonuclease activity. Plant Sci. 2002, 162, 629–636. [Google Scholar] [CrossRef]
- Qin, J.; Zhao, J.Y.; Zuo, K.J.; Cao, Y.F.; Ling, H.; Sun, X.F.; Tang, K.X. Isolation and characterization of an ERF-like gene from Gossypium barbadense. Plant Sci. 2004, 167, 1383–1389. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, X.Y.; Liang, B.G.; Li, S.W.; Wu, Z.X.; Wang, Q.H.; Leng, C.X.; Dong, J.L.; Wang, T. Expression of baculovirus anti-apoptotic genes p35 and op-iap in cotton (Gossypium hirsutum L.) enhances tolerance to Verticillium wilt. PLoS ONE 2010, 5, e14218. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Dai, X.F. Cloning and characterization of the Gossypium hirsutum major latex protein gene and functional analysis in Arabidopsis thaliana. Planta 2010, 231, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.L.; Yang, Y.W.; Chen, T.Z.; Yu, W.G.; Liu, T.L.; Li, H.J.; Fan, X.H.; Ren, Y.Z.; Shen, D.Y.; Liu, L.; et al. Island cotton GbVe1 gene encoding a receptor-like protein confers resistance to both defoliating and non-defoliating isolates of Verticillium dahliae. PLoS ONE 2012, 7, e51091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.F.; Ding, Z.G.; Ma, Q.; Zhang, G.R.; Zhang, S.L.; Li, Z.K.; Li, W.Q.; Zhang, G.Y.; Ma, Z.Y. Transcriptome profiling of Gossypium barbadense inoculated with Verticillium dahliae provides a resource for cotton improvement. BMC Genom. 2013, 14, 637. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Jiang, H.Z.; Zhu, X.Y.; Wang, W.N.; He, X.H.; Shi, Y.Z.; Yuan, Y.L.; Du, X.M.; Cai, Y.F. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genom. 2013, 14, 852. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhu, L.F.; Tu, L.L.; Liu, L.L.; Yuan, D.J.; Jin, L.; Long, L.; Zhang, X.L. Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. J. Exp. Bot. 2011, 62, 5607–5621. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.J.; Li, Y.; Han, X.L.; Shen, F.F. Genome-wide profiling of miRNAs and other small non-coding RNAs in the Verticillium dahliae-inoculated cotton roots. PLoS ONE 2012, 7, e35765. [Google Scholar] [CrossRef] [PubMed]
- He, X.H.; Sun, Q.; Jiang, H.Z.; Zhu, X.Y.; Mo, J.H.; Long, L.; Xiang, L.X.; Xie, Y.F.; Shi, Y.Z.; Yuan, Y.L.; Cai, Y.F. Identification of novel microRNAs in the Verticillium wilt-resistant upland cotton variety KV-1 by high-throughput sequencing. Springer Plus 2014, 3, 564. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.X.; Ma, Y.P.; Yang, C.L.; Zhao, P.M.; Yao, Y.; Jian, G.L.; Luo, Y.M.; Xia, G.X. Proteomic analysis of the sea-island cotton roots infected by wilt pathogen Verticillium dahliae. Proteomics 2011, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.W.; Wang, S.Z.; Liu, K.; Si, N.; Qi, F.J.; Jian, G.L. Comparative expression analysis in susceptible and resistant Gossypium hirsutum responding to Verticillium dahliae infection by cDNA-AFLP. Physiol. Mol. Plant Pathol. 2012, 80, 50–57. [Google Scholar] [CrossRef]
- Gao, W.; Long, L.; Zhu, L.; Xu, L.; Gao, W.; Sun, L.Q.; Liu, L.L.; Zhang, X.L. Proteomic and virusinduced gene silencing (VIGS) analyses reveal that gossypol, brassinosteroids and jasmonic acid contribute to the resistance of cotton to Verticillium dahliae. Mol. Cell. Proteom. 2013, 12, 3690–3703. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.B.; Wang, Z.W.; Li, F.G.; Ye, W.W.; Wang, J.Y.; Song, G.L.; Yue, Z.; Cong, L.; Shang, H.H.; Zhu, S.L.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Naoumkina, M.A.; Zhao, Q.; Gallego-Giraldo, L.; Dai, X.; Zhao, P.X.; Dixon, R.A. Genome-wide analysis of phenylpropanoid defence pathways. Mol. Plant Pathol. 2010, 11, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Smit, F.; Dubery, L.A. Cell wall reinforcement in cotton hypocotyls in response to a Verticillium dahliae elicitor. Phytochemistry 1997, 44, 811–815. [Google Scholar] [CrossRef]
- Pomar, F.; Novo, M.; Bernal, M.A.; Merino, F.; Barcelo, A.R. Changes in stem lignins (monomer composition and crosslinking) and peroxidase are related with the maintenance of leaf photosynthetic integrity during Verticillium wilt in Capsicum annuum. New Phytol. 2004, 163, 111–123. [Google Scholar] [CrossRef]
- Gayoso, C.; Pomar, F.; Novo-Uzal, E.; Merino, F.; Martinez de Ilarduya, O. The Ve-mediated resistance response of the tomato to Verticillium dahliae involves H2O2, peroxidase and lignins and drives PAL gene expression. BMC Plant Biol. 2010, 10, 232–251. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.; Singh, N.; Nandi, A.K. Identification of plant defence regulators through transcriptional profiling of Arabidopsis thaliana cdd1 mutant. J. Biosci. 2015, 40, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Matern, S.; Peskan-Berghoefer, T.; Gromes, R.; Kiesel, R.V.; Rausch, T. Imposed glutathione-mediated redox switch modulates the tobacco wound-induced protein kinase and salicylic acid induced protein kinase activation state and impacts on defence against Pseudomonas syringae. J. Exp. Bot. 2015, 66, 1935–1950. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, J.; Henderson, M.; Schweizer, P.; Burton, R.A.; Fincher, G.B.; Little, A. Differential accumulation of callose, arabinoxylan and cellulose in nonpenetrated versus penetrated papillae on leaves of barley infected with Blumeria graminis f. sp. hordei. New Phytol. 2014, 204, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.H.; Bauw, G.; Welinder, K.G.; van Montagu, M.; Boerjan, W. Purification and characterization of peroxidases correlated with lignification in poplar xylem. Plant Physiol. 1998, 118, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Ray, H.; Hammerschmidt, R. Responses of potato tuber to infection by Fusarium sambucinum. Physiol. Mol. Plant Pathol. 1998, 53, 81–92. [Google Scholar] [CrossRef]
- Treutter, D. Significance of flavonoids in plant resistance and enhancement of their biosynthesis. Plant Biol. 2005, 7, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Wildermuth, M.C.; Dewdney, J.; Wu, G.; Ausubel, F.M. Isochorismate synthase is required to synthesize salicylic acid for plant defence. Nature 2001, 414, 562–565. [Google Scholar] [PubMed]
- Jelenska, J.; Yao, N.; Vinatzer, B.A.; Wright, C.M.; Brodsky, J.L.; Greenberg, J.T. A J domain virulence effector of Pseudomonas syringae remodels host chloroplasts and suppresses defenses. Curr. Biol. 2007, 17, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, I.; Kissen, R.; Bones, A.M. Phytoalexins in defense against pathogens. Trends Plant Sci. 2012, 17, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Venisse, J.S.; Malnoy, M.; Faize, M.; Paulin, J.P.; Brisset, M.N. Modulation of defense responses of Malus spp. during compatible and incompatible interactions with Erwinia amylovora. Mol. Plant Microbe Interact. 2002, 15, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Thilmony, R.; Underwood, W.; He, S.Y. Genome-wide transcriptional analysis of the Arabidopsis thaliana interaction with the plant pathogen Pseudomonas syringae pv. tomato DC3000 and the human pathogen Escherichia coli O157:H7. Plant J. 2006, 46, 34–53. [Google Scholar] [CrossRef] [PubMed]
- Truman, W.; de Zabala, M.T.; Grant, M. Type III effectors orchestrate a complex interplay between transcriptional networks to modify basal defence responses during pathogenesis and resistance. Plant J. 2006, 46, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant. 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Bari, R.; Jones, J.D. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, C.M.; van der Does, D.; Zamioudis, C.; Leon-Reyes, A.; van Wees, S.C. Hormonal modulation of plant immunity. Annu. Rev. Cell Dev. Biol. 2012, 28, 489–521. [Google Scholar] [CrossRef] [PubMed]
- Glazebrook, J.; Chen, W.J.; Estes, B.; Chang, H.S.; Nawrath, C.; Metraux, J.P.; Zhu, T.; Katagiri, F. Topology of the network integrating salicylate and jasmonate signal transduction derived from global expression phenotyping. Plant J. 2003, 34, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Vlot, A.C.; Dempsey, D.A.; Klessig, D.F. Salicylic acid, a multi-faceted hormone to combat disease. Annu. Rev. Phytopathol. 2009, 47, 177–206. [Google Scholar] [CrossRef] [PubMed]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J. Fungal cell wall chitinases and glucanases. Microbiology 2004, 150, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, S.D.; Novak, N.G. Expression of poplar chitinase in tomato leads to inhibition of development in Colorado potato beetle. Biotechnol. Lett. 2006, 28, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Nurnberger, T.; Brunner, F.; Kemmerling, B.; Piater, L. Innate immunity in plants and animals: Striking similarities and obvious differences. Immunol. Rev. 2004, 198, 249–266. [Google Scholar] [CrossRef] [PubMed]
- Catanzariti, A.M.; Dodds, P.N.; Ve, T.; Kobe, B.; Ellis, J.G.; Staskawicz, B.J. The AvrM effector from Flax rust has a structured C-terminal domain and interacts directly with the M resistance protein. Mol. Plant-Microbe Interact. 2010, 23, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Ade, J.; de Young, B.J.; Golstein, C.; Innes, R.W. Indirect activation of a plant nucleotide binding site-leucine-rich repeat protein by a bacterial protease. Proc. Natl. Acad. Sci. USA 2007, 104, 2531–2536. [Google Scholar] [CrossRef] [PubMed]
- Zuo, K.; Wang, J.; Wu, W.; Chai, Y.; Sun, X.; Tang, K. Identification and characterization of differentially expressed ESTs of Gossypium barbadense infected by Verticillium dahliae with suppression subtractive hybridization. Mol. Biol. 2005, 39, 214–223. [Google Scholar] [CrossRef]
- Bradford, M.M. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Liu, Y.H.; Lin-Wang, K.; Deng, C.; Warran, B.; Wang, L.; Yu, B.; Yang, H.Y.; Wang, J.; Espley, R.V.; Zhang, J.L.; et al. Comparative transcriptome analysis of white and purple potato to identify genes involved in anthocyanin biosynthesis. PLoS ONE 2015, 10, e0129148. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; Yamanishi, Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, J.; Wang, Z.; Zhu, Q.; Wang, W. Hormonal changes in the grains of rice subjected to water stress during grain filling. Plant Physiol. 2001, 127, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Y.; Zhang, C.X. Data Processing System (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 2013, 20, 254–260. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, W.; Xie, D.; Zhu, H.; Li, W.; Xu, Z.; Yang, L.; Li, Z.; Sun, L.; Wang, J.; Nie, L.; et al. Comparative Proteomic Analysis of Gossypium thurberi in Response to Verticillium dahliae Inoculation. Int. J. Mol. Sci. 2015, 16, 25121-25140. https://doi.org/10.3390/ijms161025121
Fang W, Xie D, Zhu H, Li W, Xu Z, Yang L, Li Z, Sun L, Wang J, Nie L, et al. Comparative Proteomic Analysis of Gossypium thurberi in Response to Verticillium dahliae Inoculation. International Journal of Molecular Sciences. 2015; 16(10):25121-25140. https://doi.org/10.3390/ijms161025121
Chicago/Turabian StyleFang, Weiping, Deyi Xie, Heqin Zhu, Wu Li, Zhenzhen Xu, Lirong Yang, Zhifang Li, Li Sun, Jinxia Wang, Lihong Nie, and et al. 2015. "Comparative Proteomic Analysis of Gossypium thurberi in Response to Verticillium dahliae Inoculation" International Journal of Molecular Sciences 16, no. 10: 25121-25140. https://doi.org/10.3390/ijms161025121