
Evaluation of New Fluorescent Lipophosphoramidates for Gene Transfer and Biodistribution Studies after Systemic Administration

 ,
,
Abstract
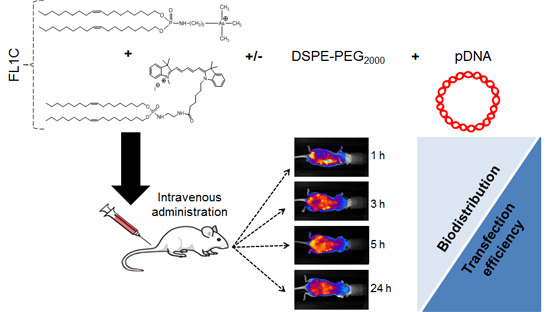
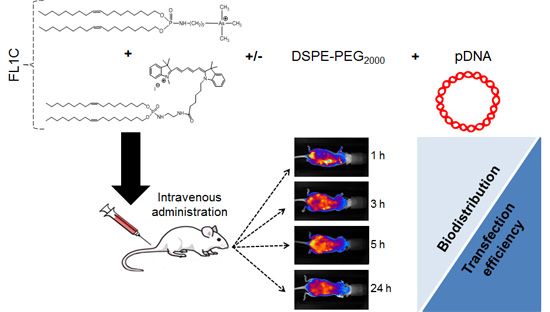
:
1. Introduction
2. Results and Discussion
2.1. Fluorescent Formulations
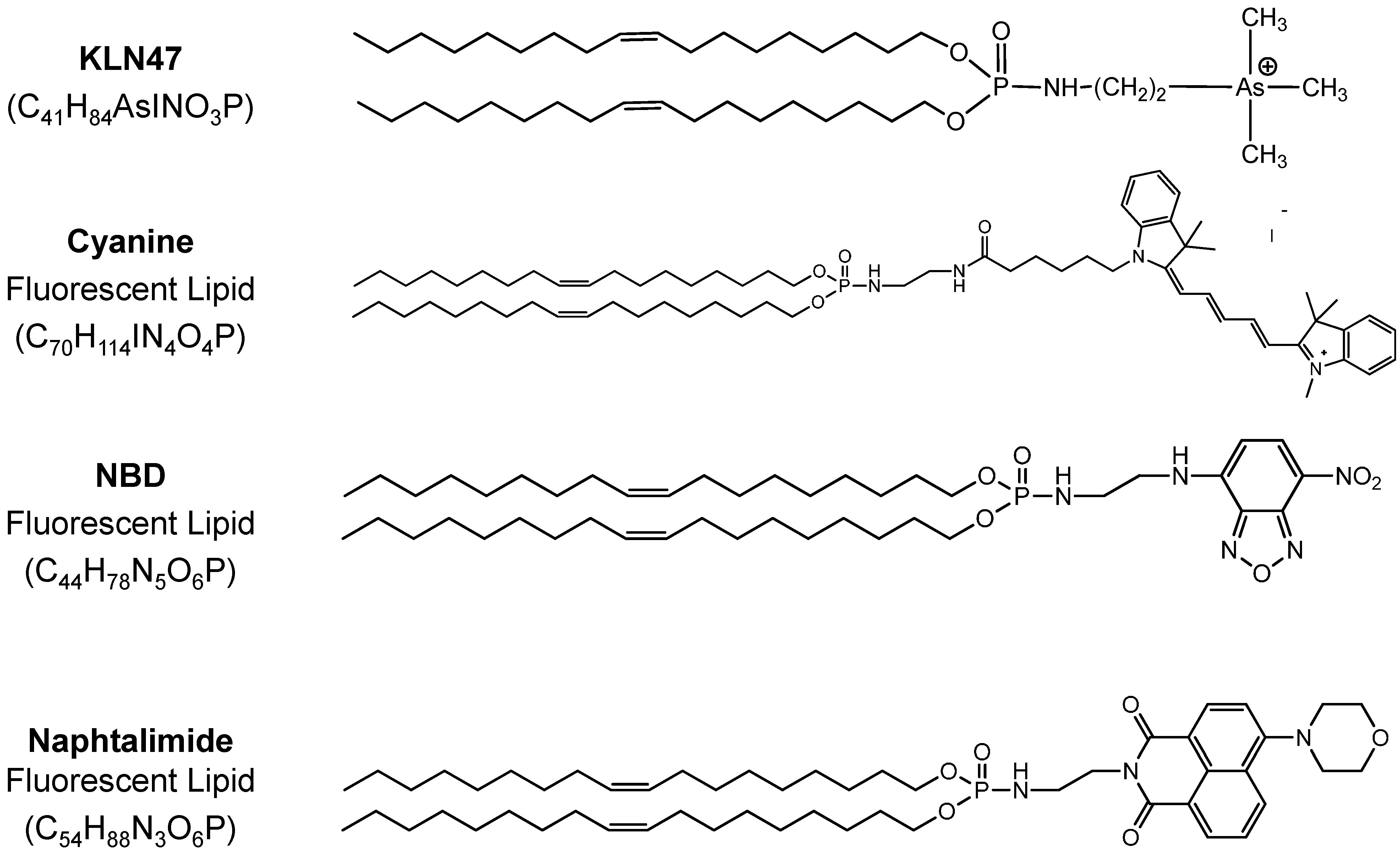

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Size (nm) | Poly Index | ζ Potential (mV) |
|---|---|---|---|
| KLN47a—1.5 mM, 1.3 mg/mL | 220.9 | - | +60.2 |
| FL1A: KLN47a—1.5 mM + FL Cyanine 5 mol % | 189 | - | +44.0 |
| FL2A: KLN47a—1.5 mM + FL NBD 5 mol % | 167.5 | 0.561 | +53.1 |
| FL2B: KLN47a—1.5 mM + FL NBD 10 mol % | 205.0 | 0.418 | +43.0 |
| FL3A: KLN47a—1.5 mM + FL Naphtalimide 5 mol % | 353.0 | 0.668 | +50.5 |
| FL3B: KLN47a—1.5 mM + FL Naphthalimide 10 mol % | 354.8 | 0.477 | +56.2 |
| KLN47b—11.5 mM, 10 mg/mL | 234.8 | 0.560 | +58.4 |
| FL1B: KLN47b—11.5 mM + FL Cyanine 5 mol % | 141.4 | 0.714 | +48.1 |
| KLN47c—23 mM, 20 mg/mL | 286.8 | 0.792 | +65.9 |
| FL1C: KLN47c—23 mM + FL Cyanine 5 mol % | 164.5 | 0.474 | +54.4 |
2.2. In Vitro Evaluation
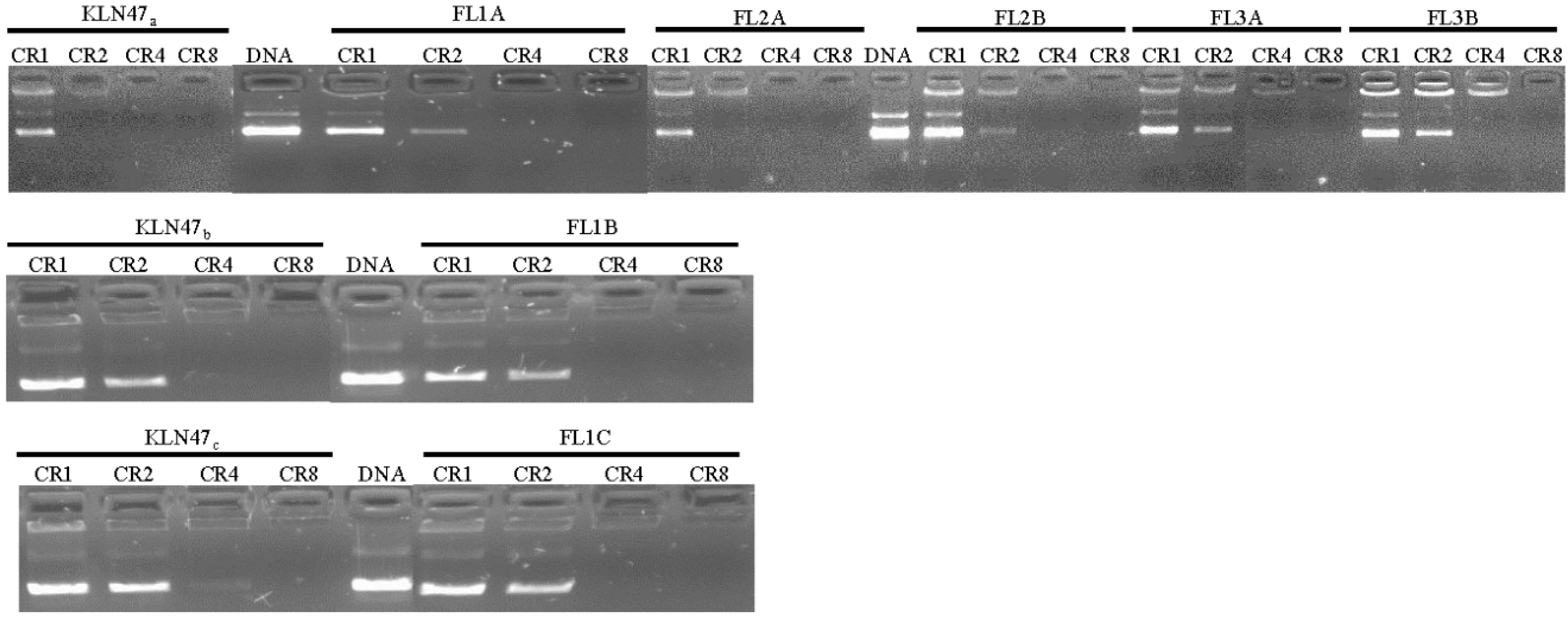
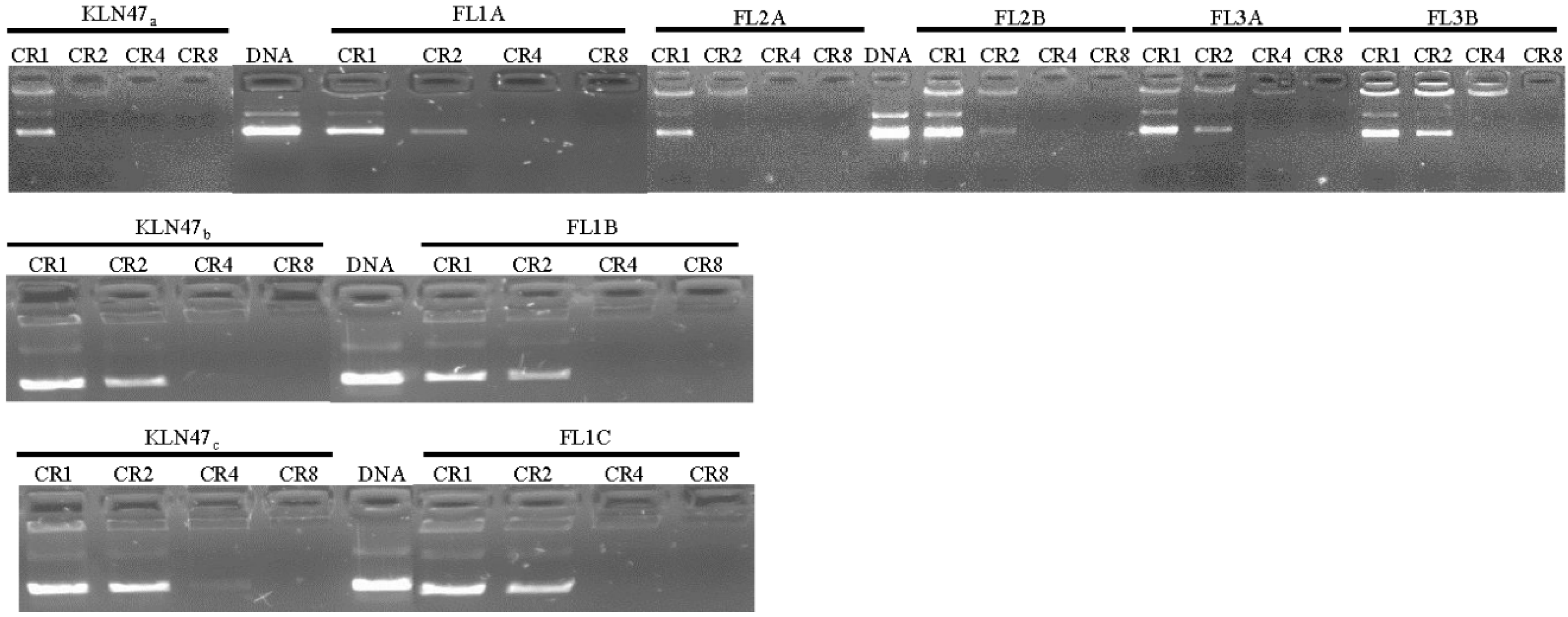
2.2.1. DNA Condensation Ability
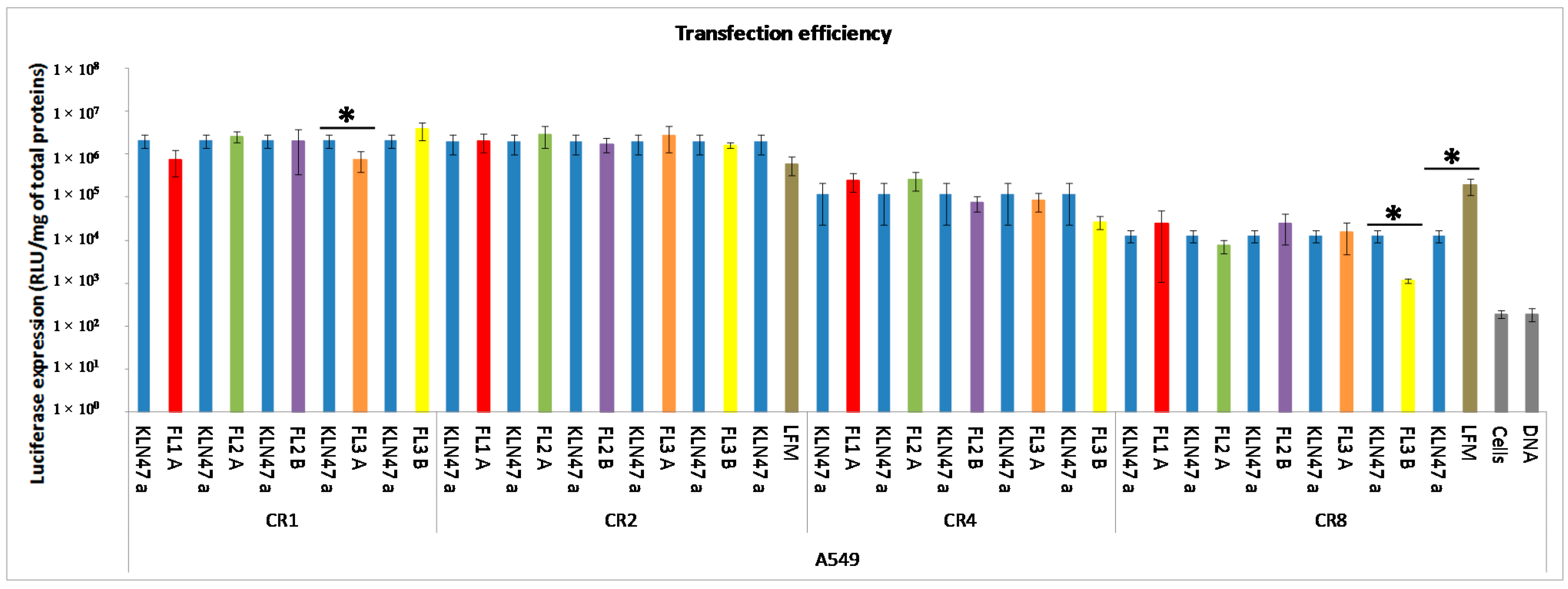
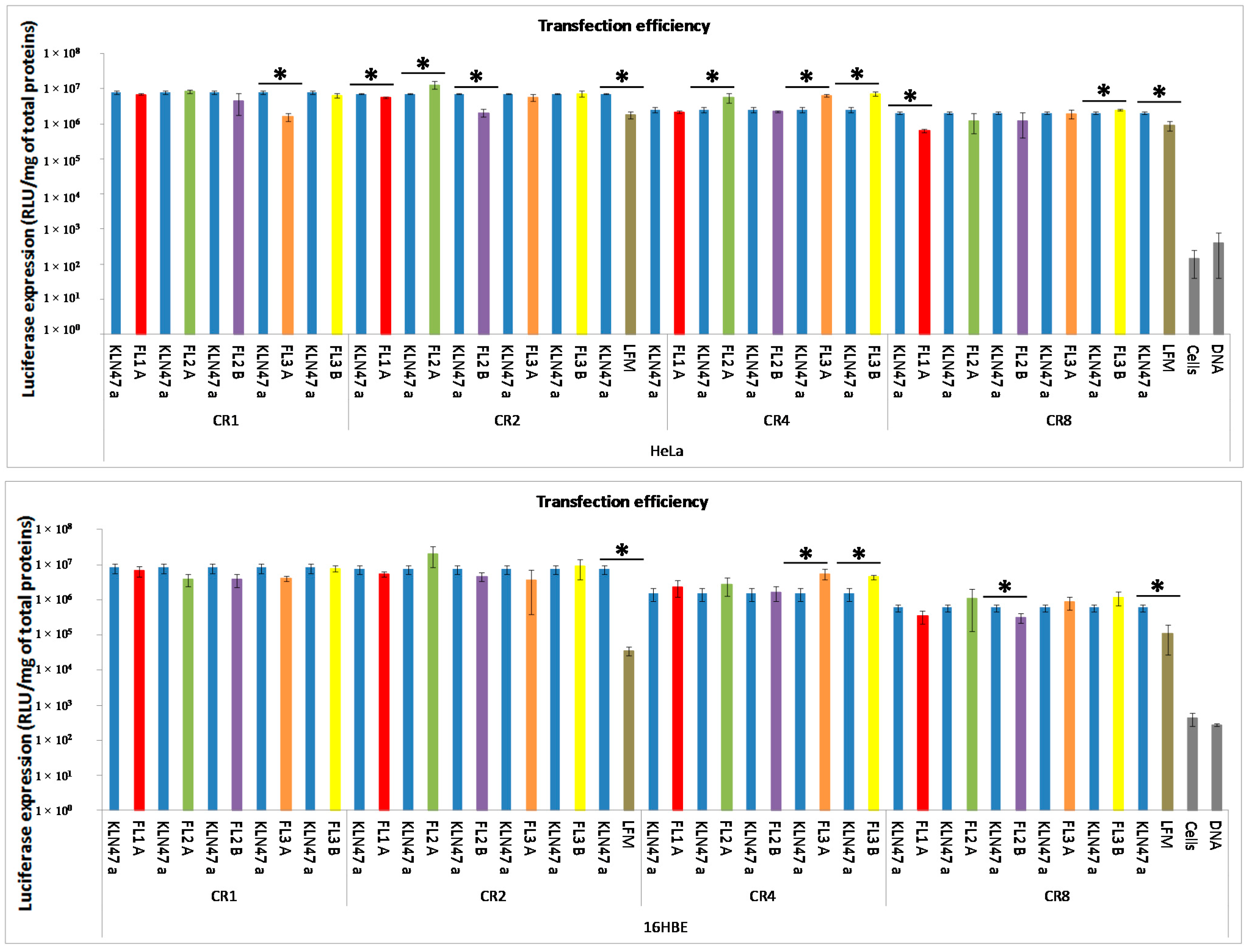
2.2.2. In Vitro Transfection Assays
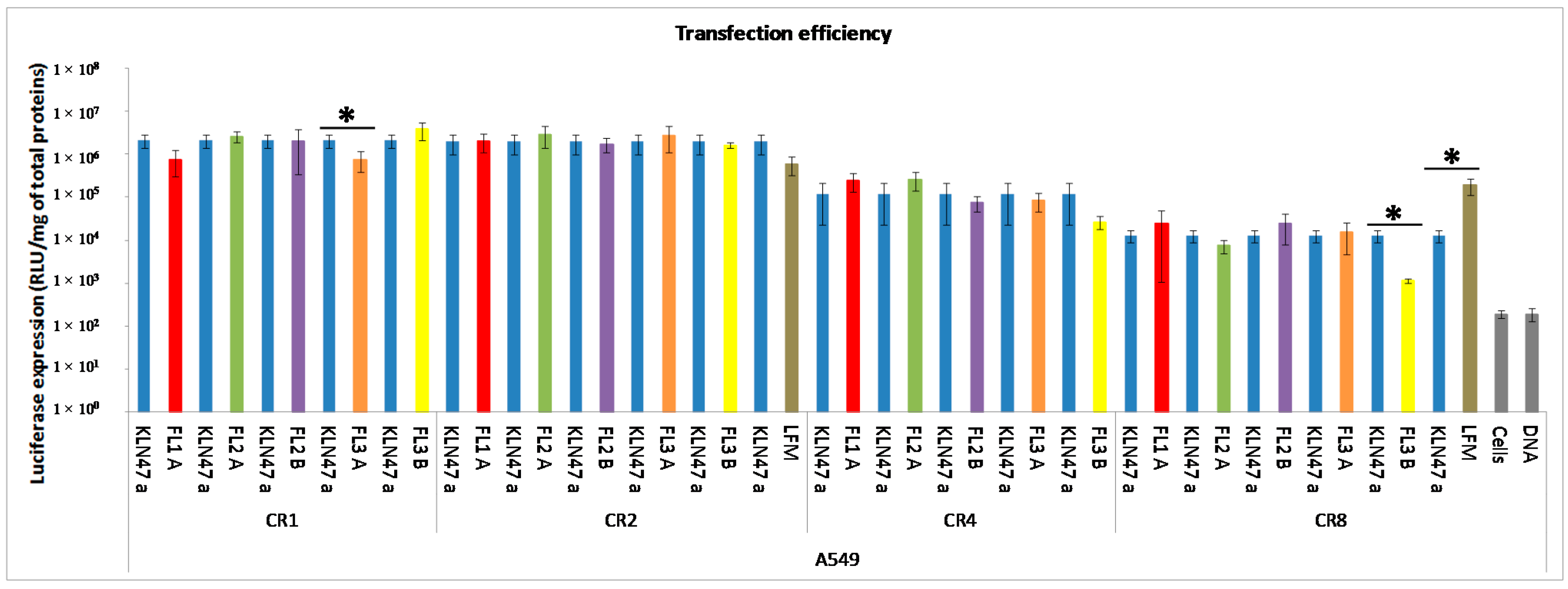
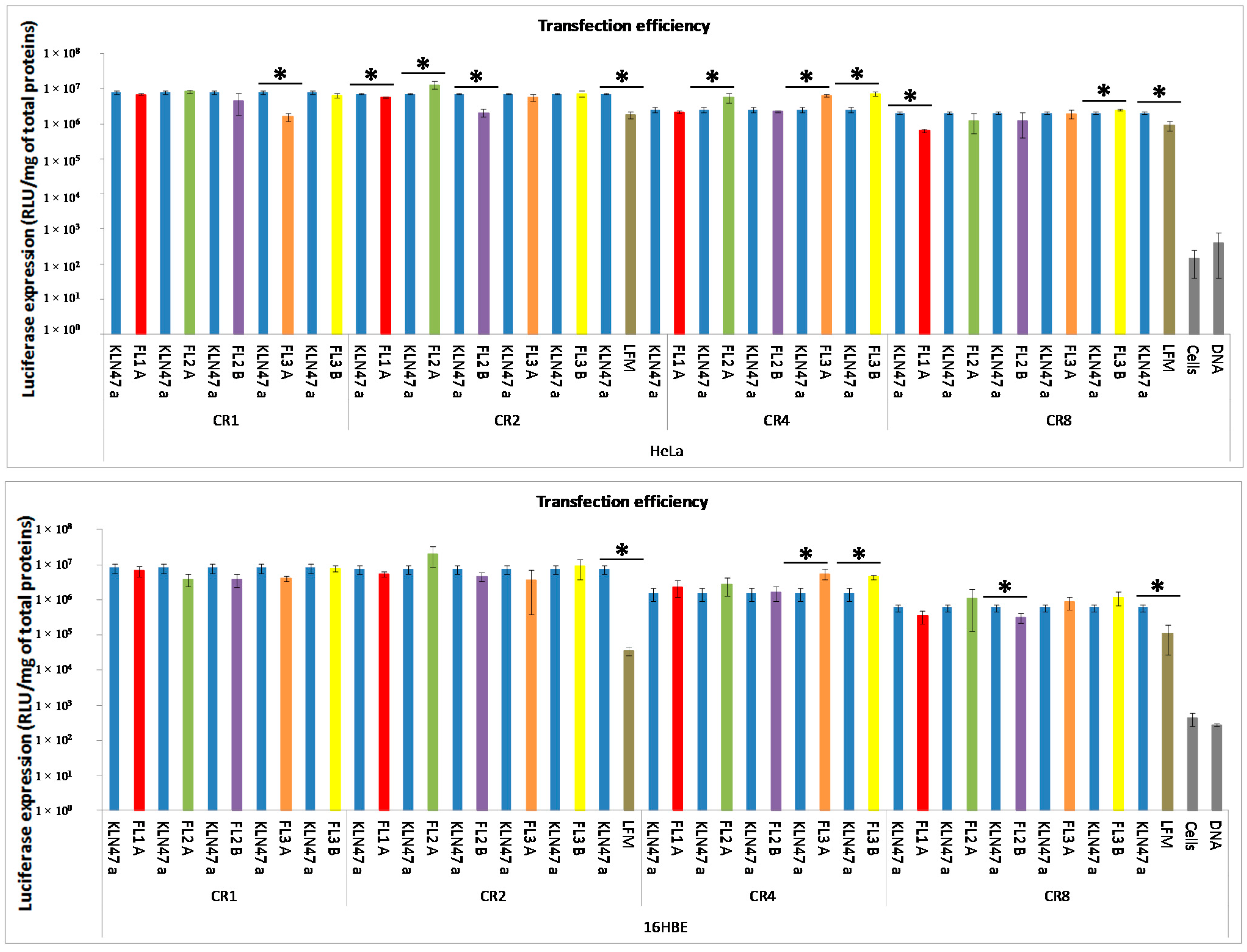
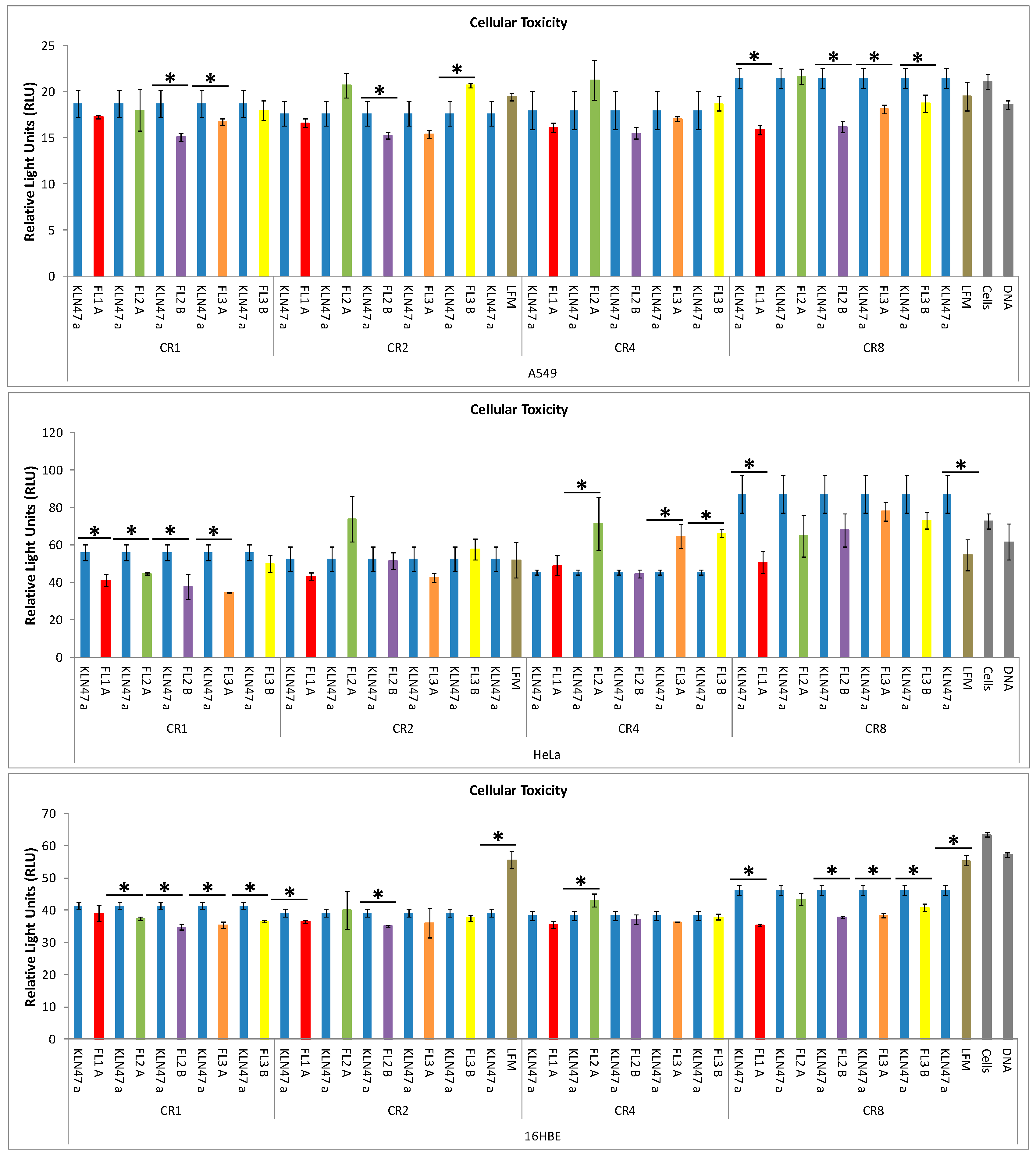
2.2.3. In Vitro Cytotoxicity
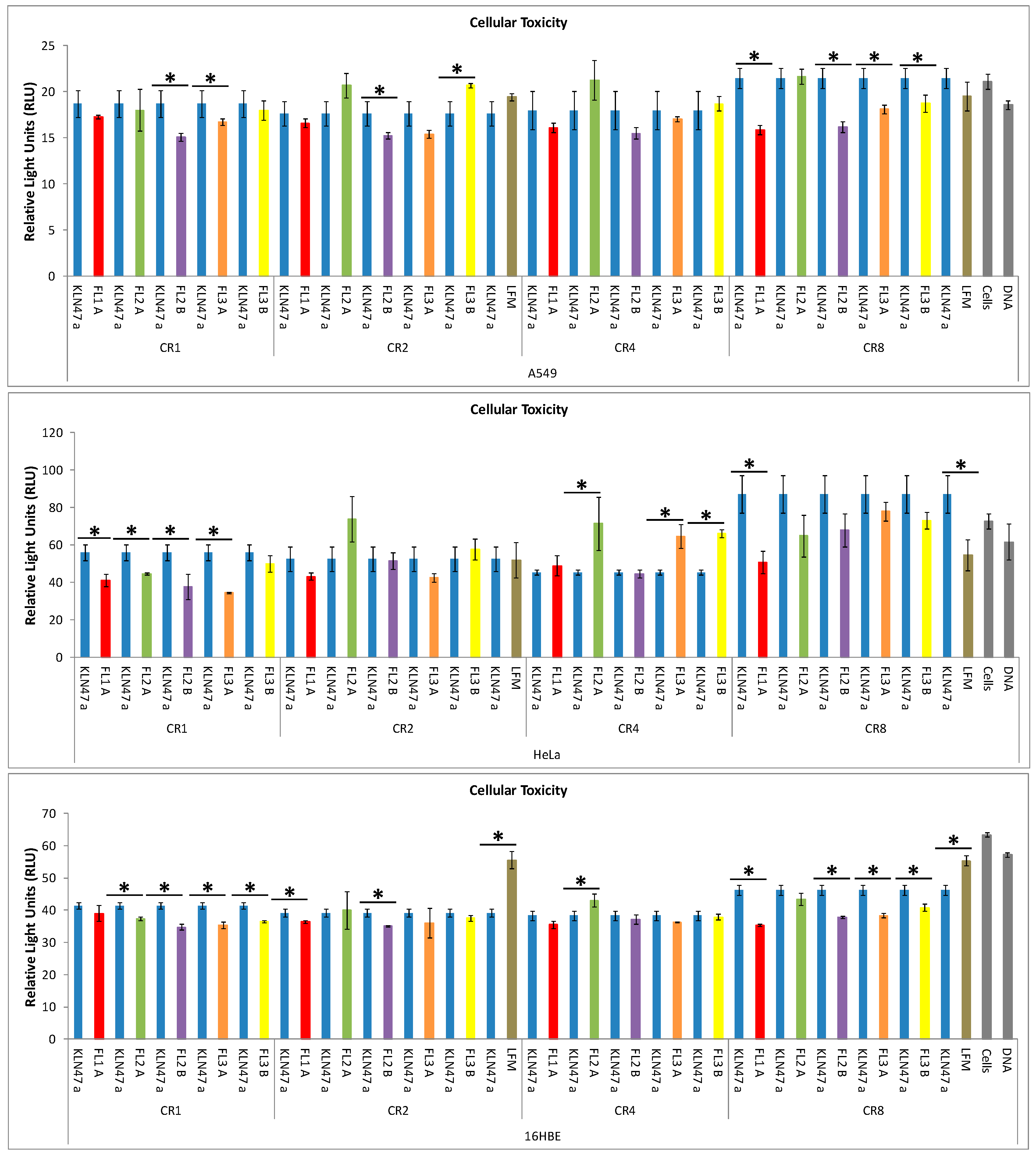
2.3. In Vivo Experiments
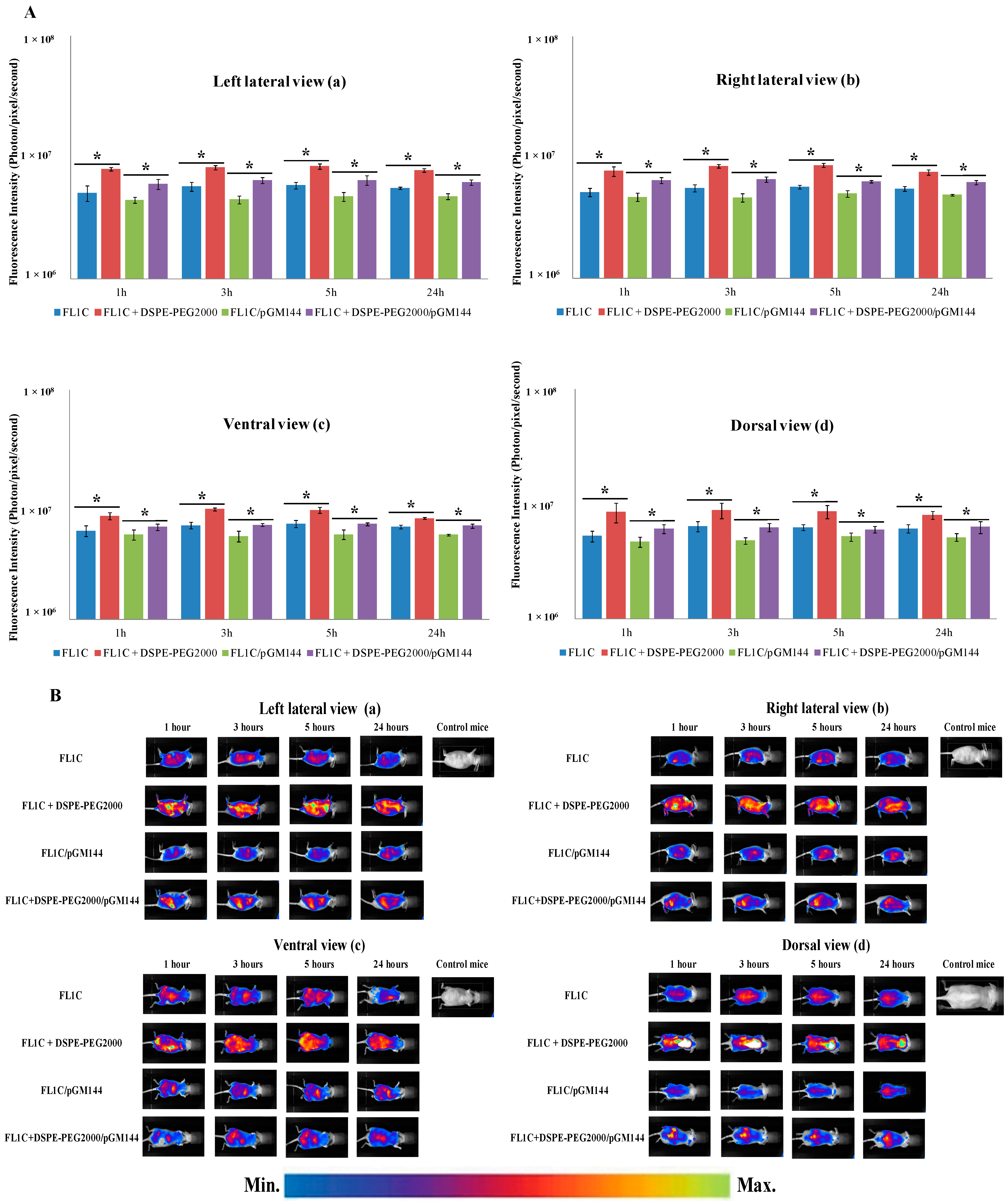
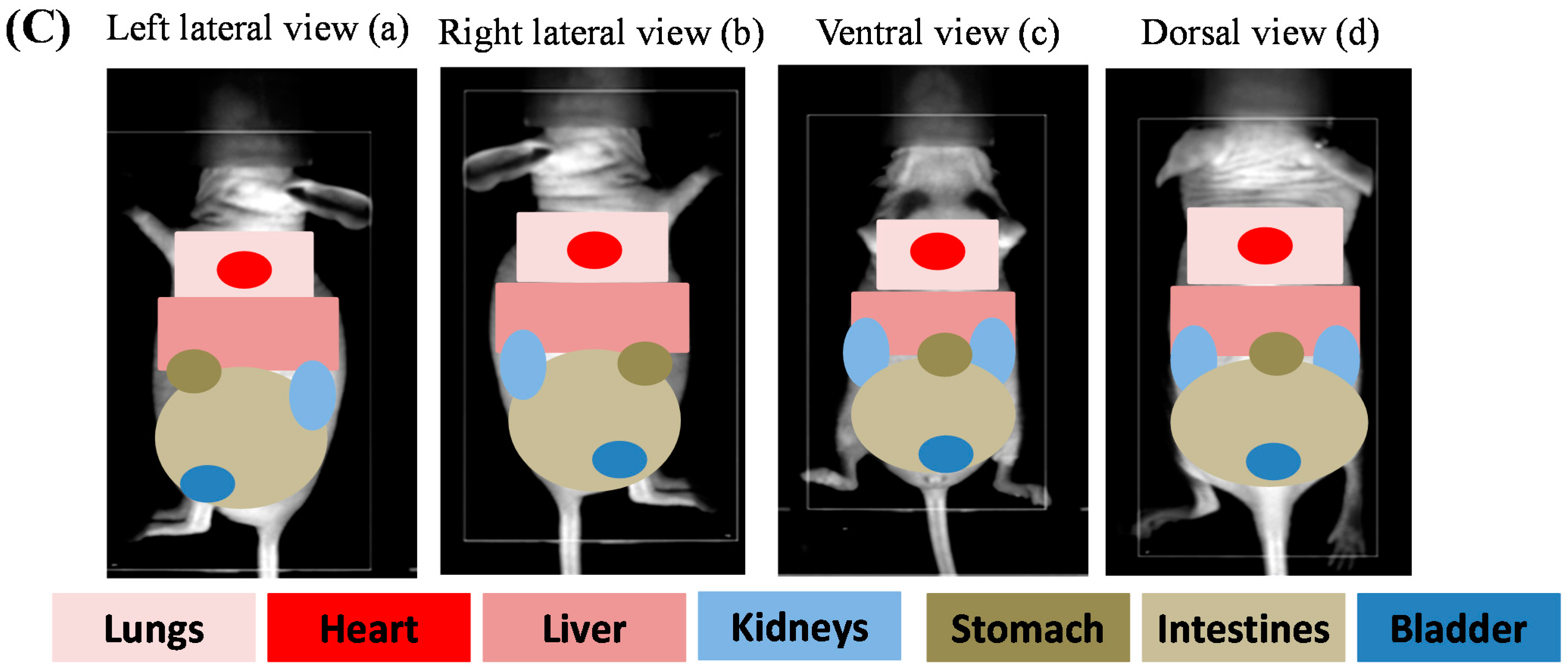
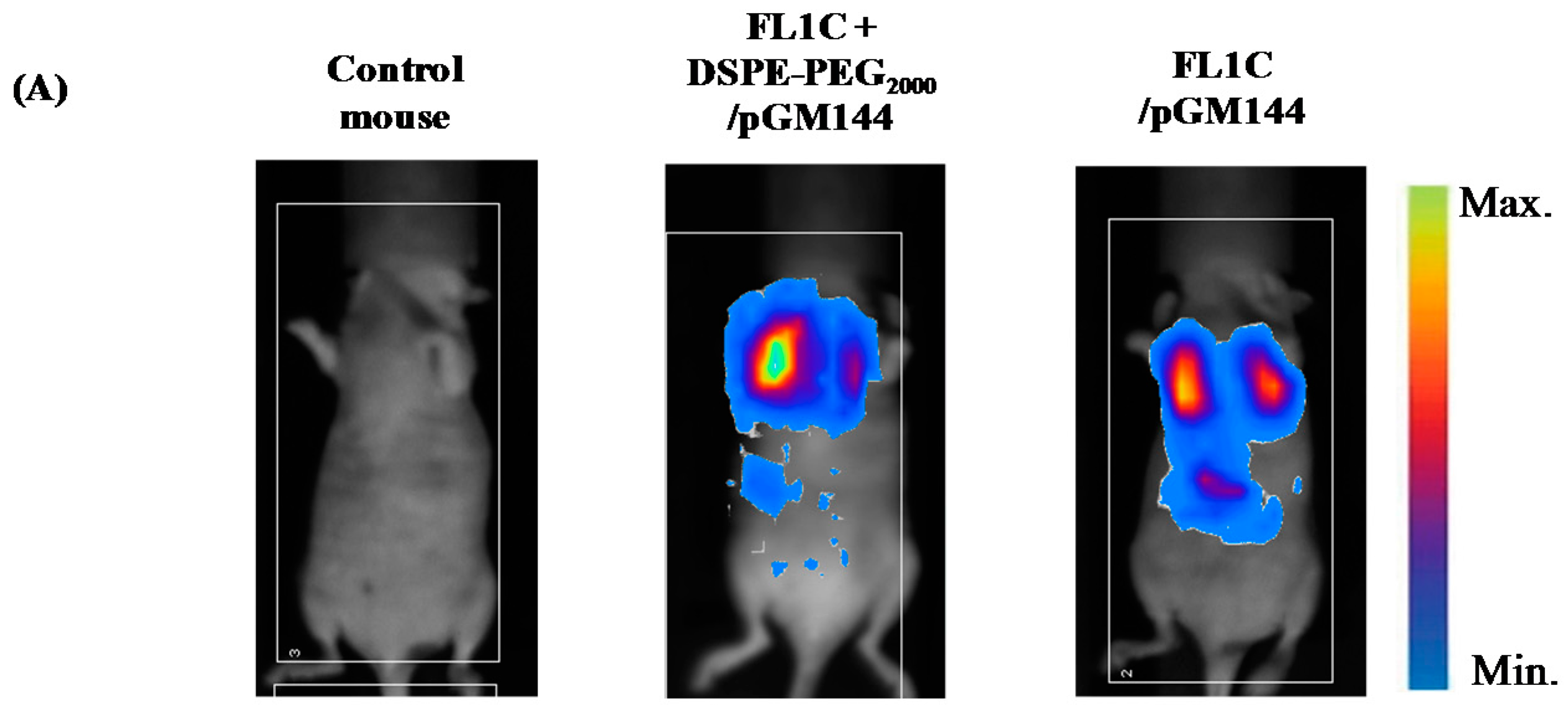
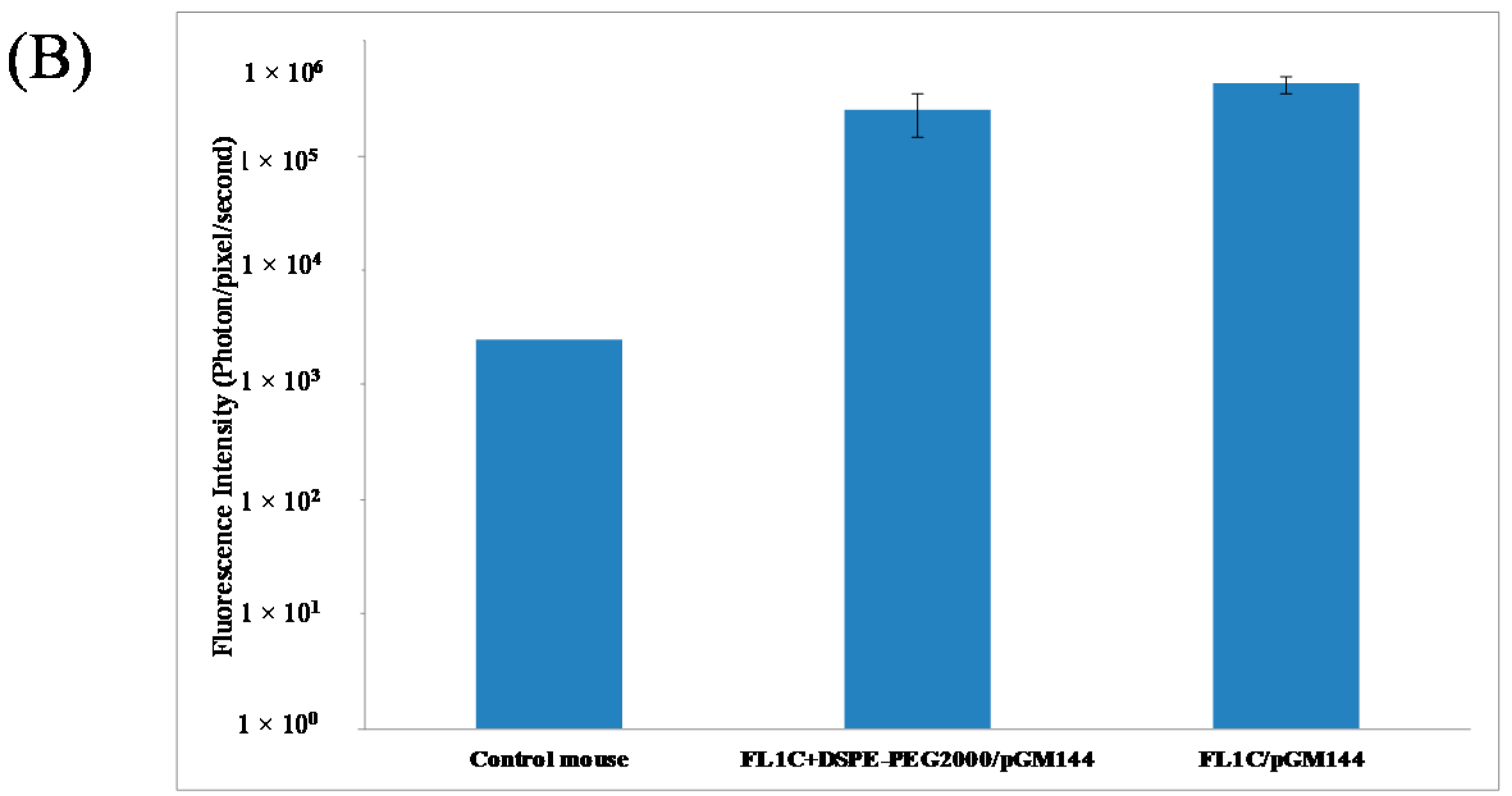
2.3.1. In Vivo Biodistribution
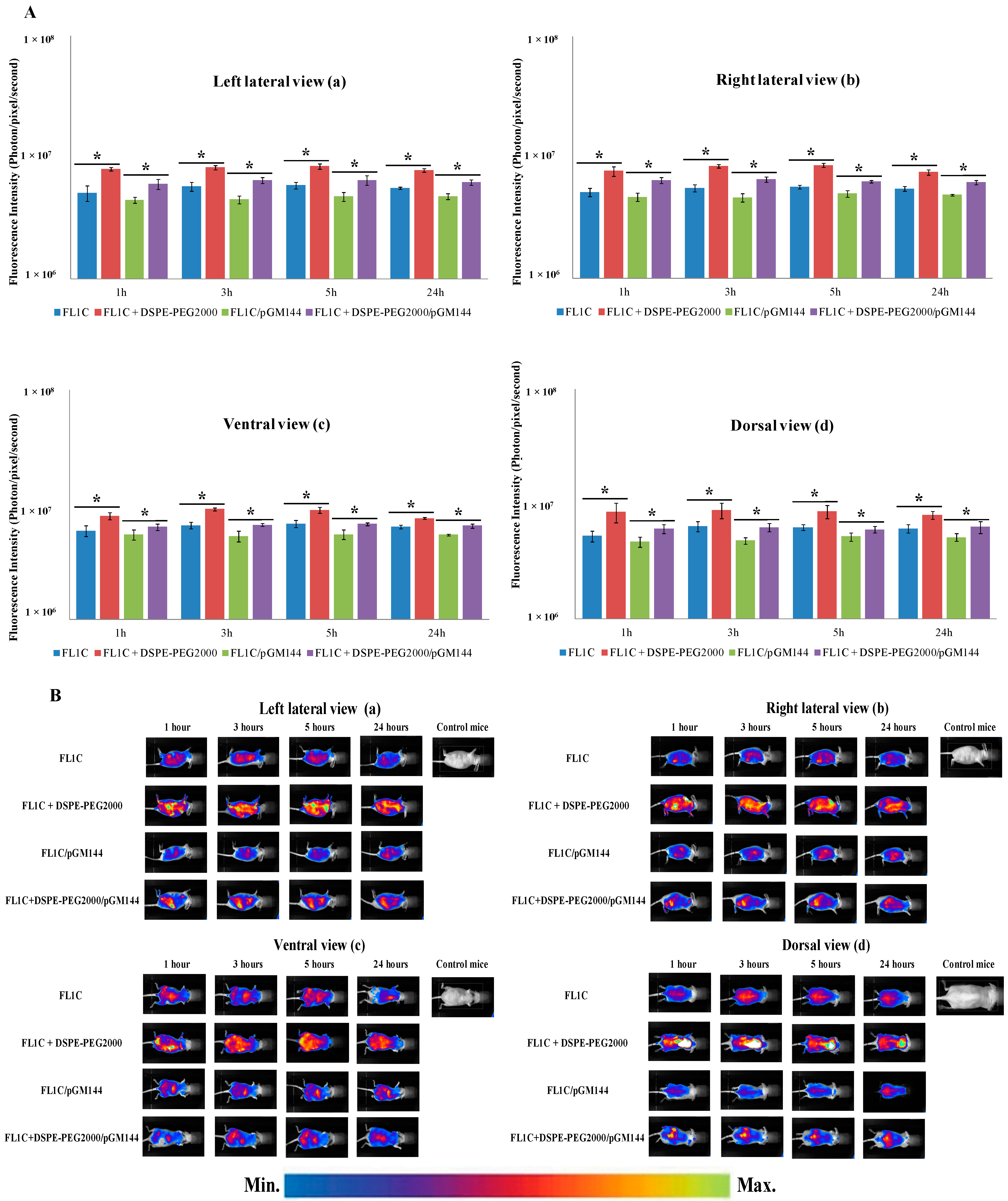

2.3.2. In Vivo Bioluminescence Imaging


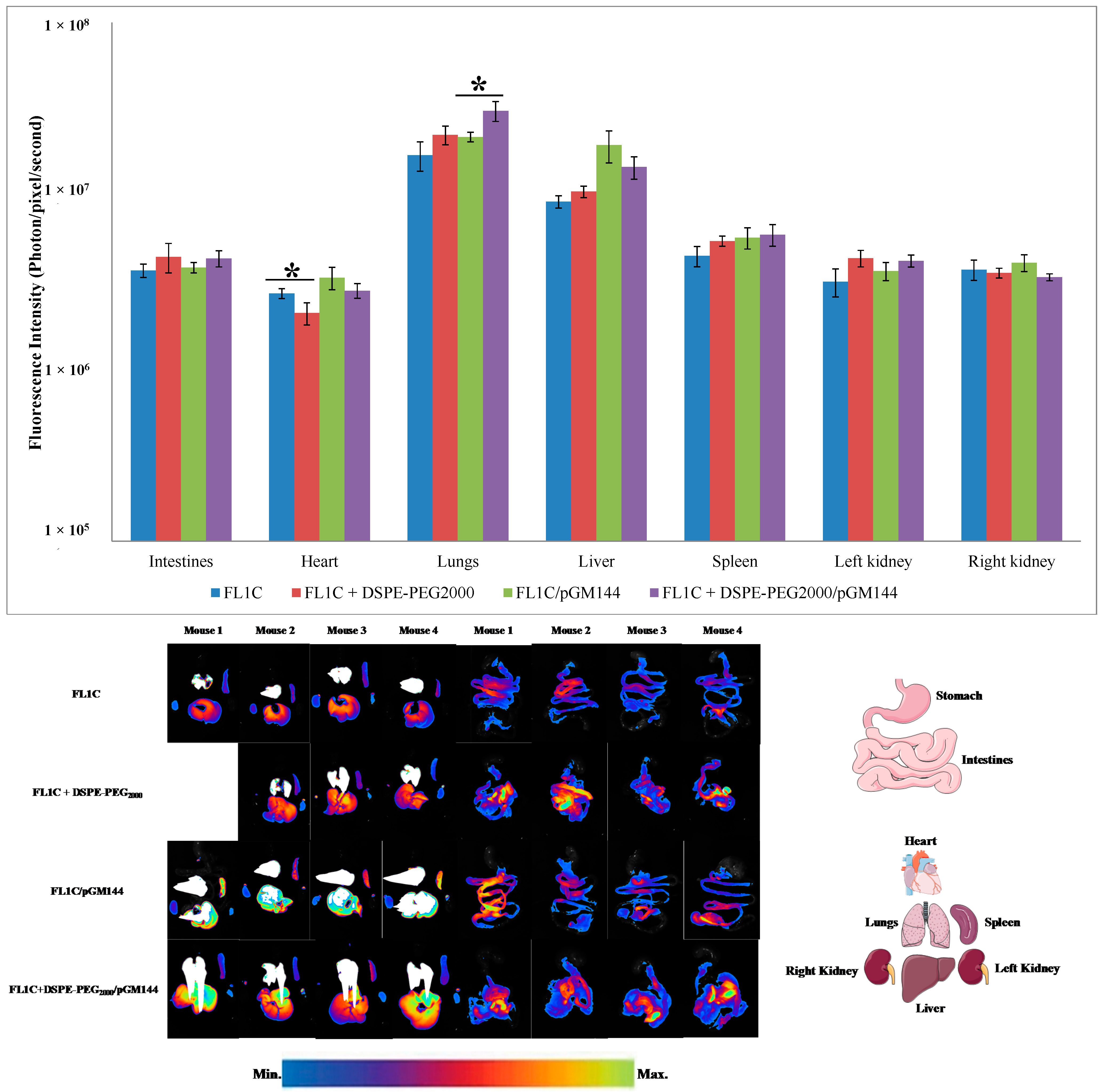
2.3.3. Biofluorescence Imaging in Isolated Organs

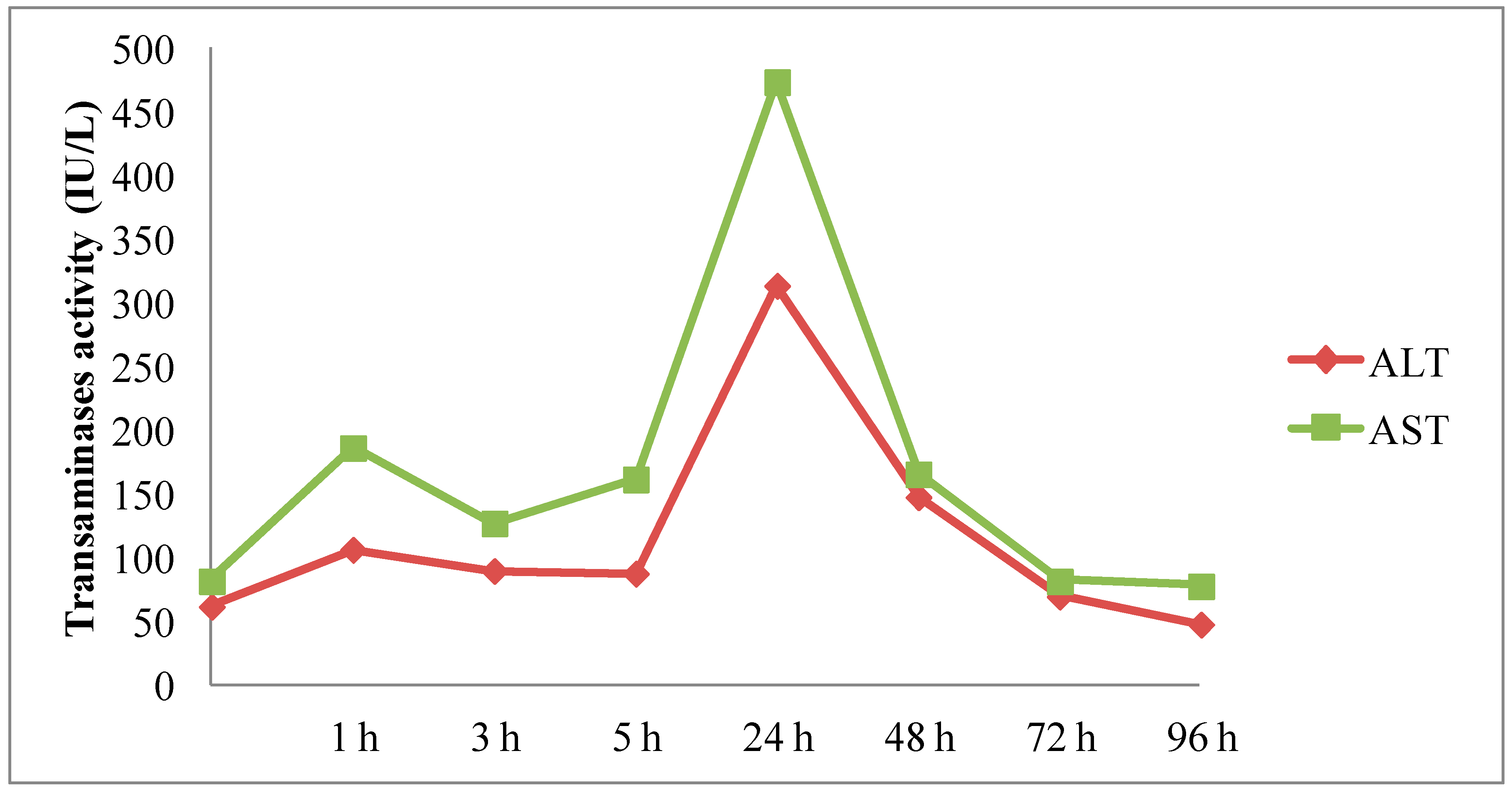
2.3.4. In Vivo Hepatotoxicity

3. Experimental Section
3.1. Formulation of Fluorescent Cationic Lipids
3.1.1. General Methods
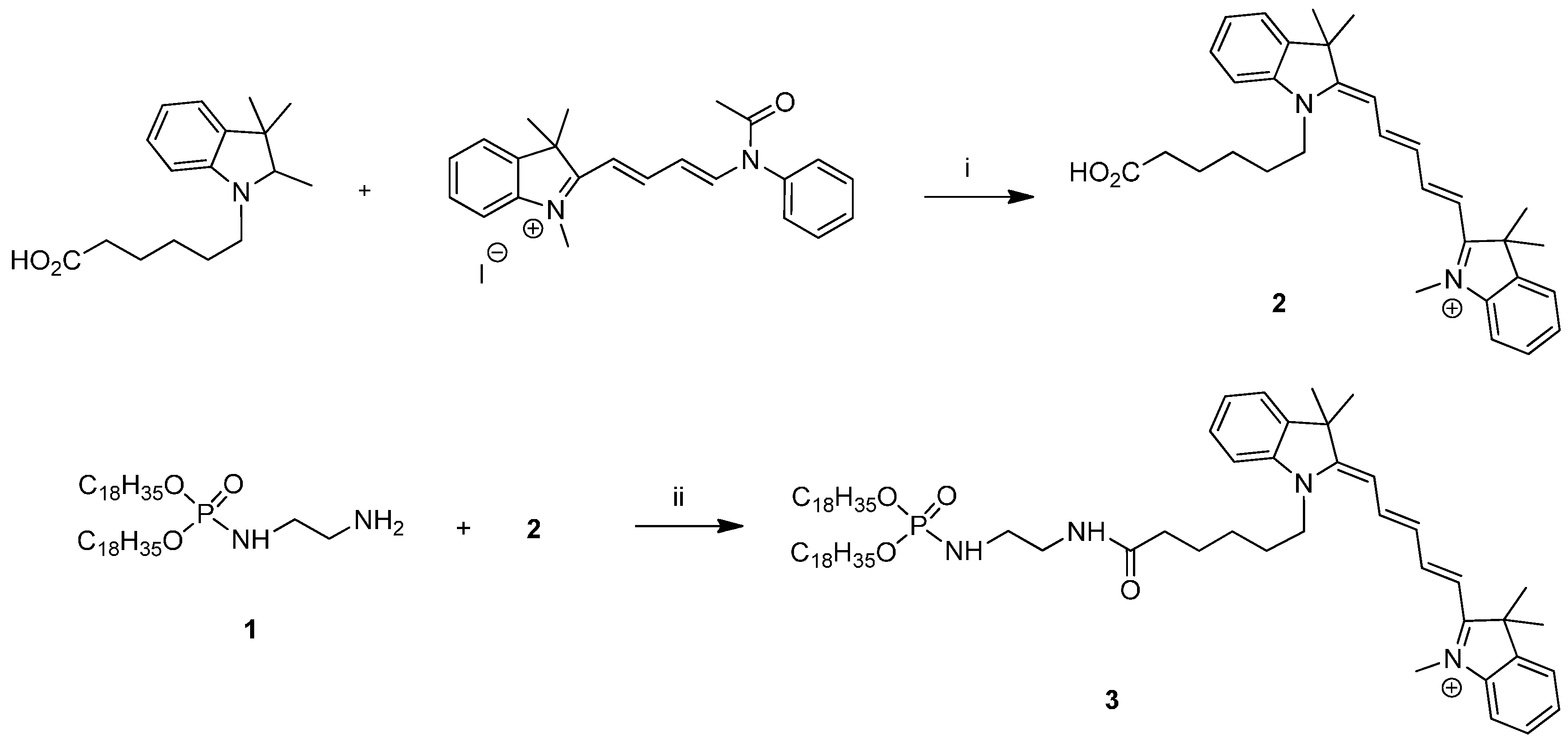
3.1.2. Absorption
3.1.3. Fluorescence
3.1.4. Liposomes
3.2. In Vitro Experiments
3.2.1. Plasmid DNA
3.2.2. DNA Condensation Ability
3.2.3. In Vitro Transfection Assays
Cell Lines
Transfection Procedure
Reporter Gene Expression
Cytotoxicity Assessment
3.3. In Vivo Experiments
3.3.1. In Vivo Systemic Administration
3.3.2. In Vivo Biofluorescence Imaging
3.3.3. In Vivo Bioluminescence Imaging
3.3.4. Biofluorescence Imaging in Isolated Organs
3.3.5. In vivo Hepatotoxicity Assessment
3.4. Statistical Test
4. Conclusion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Baker, M. Whole-animal imaging: The whole picture. Nature 2010, 463, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Goutayer, M.; Dufort, S.; Josserand, V.; Royère, A.; Heinrich, E.; Vinet, F.; Bibette, J.; Coll, J.-L.; Texier, I. Tumor targeting of functionalized lipid nanoparticles: Assessment by in vivo fluorescence imaging. Eur. J. Pharm. Biopharm. 2010, 75, 137–147. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Carmoy, N.; Resnier, P.; Denis, C.; Misery, L.; Pitard, B.; Benoit, J.-P.; Passirani, C.; Montier, T. In vivo imaging of DNA lipid nanocapsules after systemic administration in a melanoma mouse model. Int. J. Pharm. 2012, 423, 108–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, A.; Nakano, M.; Saito, K.; Haruno, R.; Watanabe, T.M.; Ohyanagi, T.; Jin, T.; Okada, Y.; Nagai, T. Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc. Natl. Acad. Sci. USA 2015, 112, 4352–4356. [Google Scholar] [CrossRef] [PubMed]
- Trupp, S.; Schweitzer, A.; Mohr, G.J. A fluorescent water-soluble naphthalimide-based receptor for saccharides with highest sensitivity in the physiological pH range. Org. Biomol. Chem. 2006, 4, 2965. [Google Scholar] [CrossRef] [PubMed]
- Black, S.L.; Stanley, W.A.; Filipp, F.V.; Bhairo, M.; Verma, A.; Wichmann, O.; Sattler, M.; Wilmanns, M.; Schultz, C. Probing lipid- and drug-binding domains with fluorescent dyes. Bioorg. Med. Chem. 2008, 16, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, O.; Gelb, M.H.; Schultz, C. Probing phospholipase A2 with fluorescent phospholipid substrates. ChemBioChem 2007, 8, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Berchel, M.; Haelters, J.-P.; Couthon-Gourvès, H.; Deschamps, L.; Midoux, P.; Lehn, P.; Jaffrès, P.-A. Modular Construction of fluorescent lipophosphoramidates by click chemistry. Eur. J. Org. Chem. 2011, 2011, 6294–6303. [Google Scholar] [CrossRef]
- Lamarche, F.; Mével, M.; Montier, T.; Burel-Deschamps, L.; Giamarchi, P.; Tripier, R.; Delépine, P.; le Gall, T.; Cartier, D.; Lehn, P.; Jaffrès, P.-A.; et al. Lipophosphoramidates as lipidic part of lipospermines for gene delivery. Bioconjug. Chem. 2007, 18, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Mohd Mustapa, M.F.; Bell, P.C.; Hurley, C.A.; Nicol, A.; Guénin, E.; Sarkar, S.; Writer, M.J.; Barker, S.E.; Wong, J.B.; Pilkington-Miksa, M.A.; et al. Biophysical characterization of an integrin-targeted lipopolyplex gene delivery vector. Biochemistry 2007, 46, 12930–12944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-X.; McIntosh, T.J.; Grinstaff, M.W. Functional lipids and lipoplexes for improved gene delivery. Biochimie 2012, 94, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Montier, T.; Benvegnu, T.; Jaffres, P.-A.; Yaouanc, J.-J.; Lehn, P. Progress in cationic lipid-mediated gene transfection: A series of bio-inspired lipids as an example. Curr. Gene Ther. 2008, 8, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, T.; Loizeau, D.; Picquet, E.; Carmoy, N.; Yaouanc, J.-J.; Burel-Deschamps, L.; Deleépine, P.; Giamarchi, P.; Jaffreès, P.-A.; Lehn, P.; et al. A novel cationic lipophosphoramide with diunsaturated lipid chains: Synthesis, physicochemical properties, and transfection activities. J. Med. Chem. 2010, 53, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.W.F.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef]
- Guénin, E.; Hervé, A.-C.; Floch, V.; Loisel, S.; Yaouanc, J.-J.; Clément, J.-C.; Férec, C.; des Abbayes, H. Cationic phosphonolipids containing quaternary phosphonium and arsonium groups for DNA transfection with good efficiency and low cellular toxicity. Angew. Chem. Int. Ed. 2000, 39, 629–631. [Google Scholar] [CrossRef]
- Floch, V.; Delépine, P.; Guillaume, C.; Loisel, S.; Chassé, S.; le Bolc’h, G.; Gobin, E.; Leroy, J.P.; Férec, C. Systemic administration of cationic phosphonolipids/DNA complexes and the relationship between formulation and lung transfection efficiency. BBA Biomembr. 2000, 1464, 95–103. [Google Scholar] [CrossRef]
- Laurent, V.; Fraix, A.; Montier, T.; Cammas-Marion, S.; Ribault, C.; Benvegnu, T.; Jaffres, P.-A.; Loyer, P. Highly efficient gene transfer into hepatocyte-like HepaRG cells: New means for drug metabolism and toxicity studies. Biotechnol. J. 2010, 5, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berchel, M.; Le Gall, T.; Couthon-Gourvès, H.; Haelters, J.-P.; Montier, T.; Midoux, P.; Lehn, P.; Jaffrès, P.-A. Lipophosphonate/lipophosphoramidates: A family of synthetic vectors efficient for gene delivery. Biochimie 2012, 94, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Mével, M.; Breuzard, G.; Yaouanc, J.-J.; Clément, J.-C.; Lehn, P.; Pichon, C.; Jaffrès, P.-A.; Midoux, P. Synthesis and transfection activity of new cationic phosphoramidate lipids: High efficiency of an imidazolium derivative. ChemBioChem 2008, 9, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, S.S.; Berchel, M.; Belmadi, N.; Denis, C.; Haelters, J.-P.; Le Gall, T.; Lehn, P.; Montier, T.; Jaffrès, P.-A. Cationic lipophosphoramidates with two different lipid chains: Synthesis and evaluation as gene carriers. Org. Biomol. Chem. 2014, 12, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Berchel, M.; Le Gall, T.; Haelters, J.-P.; Lehn, P.; Montier, T.; Jaffrès, P.-A. Cationic lipophosphoramidates containing a hydroxylated polar headgroup for improving gene delivery. Mol. Pharm. 2015, 12, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.K.; Liu, F.; Chu, S.; Liu, D. Characterization of cationic liposome-mediated gene transfer in vivo by intravenous administration. Hum. Gene Ther. 1997, 8, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Orson, F.M.; Song, L.; Gautam, A.; Densmore, C.L.; Bhogal, B.S.; Kinsey, B.M. Gene delivery to the lung using protein/polyethylenimine/plasmid complexes. Gene Ther. 2002, 9, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Maiwald, G.; Haase, R.; Rogach, A.L.; Wagner, E.; de Ilarduya, C.T.; Ogris, M. Low generation PAMAM dendrimer and CpG free plasmids allow targeted and extended transgene expression in tumors after systemic delivery. J. Control. Release 2010, 146, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Nakamura, A.; Arai, S.; Kawano, K.; Maitani, Y.; Yonemochi, E. siRNA delivery to lung-metastasized tumor by systemic injection with cationic liposomes. J. Liposome Res. 2014, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.F.; Carmoy, N.; Le Gall, T.; Fraix, A.; Berchel, M.; Lorilleux, C.; Couthon-Gourvès, H.; Bellaud, P.; Fautrel, A.; Jaffrès, P.-A.; Lehn, P.; Montier, T. The gene transfection properties of a lipophosphoramidate derivative with two phytanyl chains. Biomaterials 2012, 33, 6240–6253. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.F.; Le Gall, T.; Carmoy, N.; Berchel, M.; Hyde, S.C.; Gill, D.R.; Jaffrès, P.-A.; Lehn, P.; Montier, T. Efficient in vivo transfection and safety profile of a CpG-free and codon optimized luciferase plasmid using a cationic lipophosphoramidate in a multiple intravenous administration procedure. Biomaterials 2015, 59, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hyde, S.C.; Pringle, I.A.; Abdullah, S.; Lawton, A.E.; Davies, L.A.; Varathalingam, A.; Nunez-Alonso, G.; Green, A.-M.; Bazzani, R.P.; Sumner-Jones, S.G.; et al. CpG-free plasmids confer reduced inflammation and sustained pulmonary gene expression. Nat. Biotechnol. 2008, 26, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.A.; Hyde, S.C.; Nunez-Alonso, G.; Bazzani, R.P.; Harding-Smith, R.; Pringle, I.A.; Lawton, A.E.; Abdullah, S.; Roberts, T.C.; McCormick, D.; et al. The use of CpG-free plasmids to mediate persistent gene expression following repeated aerosol delivery of pDNA/PEI complexes. Biomaterials 2012, 33, 5618–5627. [Google Scholar] [CrossRef] [PubMed]
- Belmadi, N.; Midoux, P.; Loyer, P.; Passirani, C.; Pichon, C.; le Gall, T.; Jaffrès, P.-A.; Lehn, P. Tristan Montier Synthetic vectors for gene delivery: An overview of their evolution depending on routes of administration. Biotechnol. J. 2015, 10, 1370–1389. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Pichon, C.; Yaouanc, J.-J.; Jaffrès, P.-A. Chemical vectors for gene delivery: a current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers: Histidine-rich nucleic acids carriers. Br. J. Pharmacol. 2009, 157, 166–178. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, G.; Davidson, H.; Holder, E.; Davies, L.A.; Pringle, I.A.; Sumner-Jones, S.G.; Baker, A.; Tennant, P.; Gordon, C.; Vrettou, C.; et al. Pre-clinical evaluation of three non-viral gene transfer agents for cystic fibrosis after aerosol delivery to the ovine lung. Gene Ther. 2011, 18, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Delépine, P.; Guillaume, C.; Montier, T.; Clément, J.C.; Yaouanc, J.J.; Des Abbayes, H.; Berthou, F.; le Pape, A.; Ferec, C. Biodistribution study of phosphonolipids: A class of non-viral vectors efficient in mice lung-directed gene transfer. J. Gene Med. 2003, 5, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Osaka, G.; Carey, K.; Cuthbertson, A.; Godowski, P.; Patapoff, T.; Ryan, A.; Gadek, T.; Mordenti, J. Pharmacokinetics, tissue distribution, and expression efficiency of plasmid [33P]DNA following intravenous administration of DNA/Cationic lipid complexes in mice: Use of a novel radionuclide approach. J. Pharm. Sci. 1996, 85, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Berchel, M.; Haelters, J.-P.; Afonso, D.; Maroto, A.; Deschamps, L.; Giamarchi, P.; Jaffrès, P.-A. Functionalized phospholipid molecular platform: use for production of cationic fluorescent lipids: Phospholipid molecular platform for cationic fluorescent lipids. Eur. J. Org. Chem. 2014, 2014, 1076–1083. [Google Scholar] [CrossRef]
- Davies, L.A.; McLachlan, G.; Sumner-Jones, S.G.; Ferguson, D.; Baker, A.; Tennant, P.; Gordon, C.; Vrettou, C.; Baker, E.; Zhu, J.; et al. Enhanced Lung Gene Expression After Aerosol Delivery of Concentrated pDNA/PEI Complexes. Mol. Ther. 2008, 16, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Passirani, C.; Carmoy, N.; Morille, M.; Mevel, M.; Chatin, B.; Benoit, J.-P.; Montier, T.; Pitard, B. DNA nanocarriers for systemic administration: characterization and in vivo bioimaging in healthy mice. Mol. Ther. Nucleic Acids 2013, 2, e64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Patel, A.; Spina, D.; Riffo-Vasquez, Y.; Babin-Morgan, A.; de Rosales, R.T.M.; Sunassee, K.; Clark, S.; Collins, H.; Bruce, K.; et al. In vivo biocompatibility, clearance, and biodistribution of albumin vehicles for pulmonary drug delivery. J. Control. Release 2015, 210, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gjetting, T.; Arildsen, N.S.; Christensen, C.L.; Poulsen, T.T.; Roth, J.A.; Handlos, V.N.; Poulsen, H.S. In vitro and in vivo effects of polyethylene glycol (PEG)-modified lipid in DOTAP/cholesterol-mediated gene transfection. Int. J. Nanomed. 2010, 5, 371–383. [Google Scholar]
- Le Guennic, B.; Jacquemin, D. Taking up the cyanine challenge with quantum tools. Acc. Chem. Res. 2015, 48, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Mosqueira, V.C.F.; Legrand, P.; Gulik, A.; Bourdon, O.; Gref, R.; Labarre, D.; Barratt, G. Relationship between complement activation, cellular uptake and surface physicochemical aspects of novel PEG-modified nanocapsules. Biomaterials 2001, 22, 2967–2979. [Google Scholar] [CrossRef]
- Morille, M.; Montier, T.; Legras, P.; Carmoy, N.; Brodin, P.; Pitard, B.; Benoît, J.-P.; Passirani, C. Long-circulating DNA lipid nanocapsules as new vector for passive tumor targeting. Biomaterials 2010, 31, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikram, M.; Lee, M.; Chang, C.-W.; Janát-Amsbury, M.-M.; Kern, S.E.; Kim, S.W. Long-circulating DNA-complexed biodegradable multiblock copolymers for gene delivery: Degradation profiles and evidence of dysopsonization. J. Control. Release 2005, 103, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Hirsjärvi, S.; Dufort, S.; Gravier, J.; Texier, I.; Yan, Q.; Bibette, J.; Sancey, L.; Josserand, V.; Passirani, C.; Benoit, J.-P.; et al. Influence of size, surface coating and fine chemical composition on the in vitro reactivity and in vivo biodistribution of lipid nanocapsules versus lipid nanoemulsions in cancer models. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mérian, J.; Boisgard, R.; Bayle, P.-A.; Bardet, M.; Tavitian, B.; Texier, I. Comparative biodistribution in mice of cyanine dyes loaded in lipid nanoparticles. Eur. J. Pharm. Biopharm. 2015, 93, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liggitt, D. Cationic liposome-mediated intravenous gene delivery. J. Biol. Chem. 1995, 270, 24864–24870. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Liggitt, D.; Liu, Y.; Debs, R. Systemic gene expression after intravenous DNA delivery into adult mice. Science 1993, 261, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Lunardi-Iskandar, Y.; Bryant, J.L.; Rabinovich, P.; Gallo, R.C.; Mahan, L.C. Systemic gene therapy: Biodistribution and long-term expression of a transgene in mice. Proc. Natl. Acad. Sci. USA 1995, 92, 9742–9746. [Google Scholar] [CrossRef] [PubMed]
- Kutscher, H.L.; Chao, P.; Deshmukh, M.; Singh, Y.; Hu, P.; Joseph, L.B.; Reimer, D.C.; Stein, S.; Laskin, D.L.; Sinko, P.J. Threshold size for optimal passive pulmonary targeting and retention of rigid microparticles in rats. J. Control. Release 2010, 143, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Ogris, M.; Brunner, S.; Schuller, S.; Kircheis, R.; Wagner, E. PEGylated DNA/transferrin-PEI complexes: Reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther. 1999, 6, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, T.; Barbeau, J.; Barrier, S.; Berchel, M.; Lemiègre, L.; Jeftić, J.; Meriadec, C.; Artzner, F.; Gill, D.R.; Hyde, S.C.; et al. Effects of a novel archaeal tetraether-based colipid on the in vivo gene transfer activity of two cationic amphiphiles. Mol. Pharm. 2014, 11, 2973–2988. [Google Scholar] [CrossRef] [PubMed]
- Loisel, S.; Le Gall, C.; Doucet, L.; Ferec, C.; Floch, V. Contribution of plasmid DNA to hepatotoxicity after systemic administration of lipoplexes. Hum. Gene Ther. 2001, 12, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kawakami, S.; Charoensit, P.; Higuchi, Y.; Hashida, M. Evaluation of proinflammatory cytokine production and liver injury induced by plasmid DNA/cationic liposome complexes with various mixing ratios in mice. Eur. J. Pharm. Biopharm. 2009, 71, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Kako, K.; Nishikawa, M.; Yoshida, H.; Takakura, Y. Effects of inflammatory response on in vivo transgene expression by plasmid DNA in mice. J. Pharm. Sci. 2008, 97, 3074–3083. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, S.; Ito, Y.; Fumoto, S.; Yamashita, F.; Hashida, M. Enhanced gene expression in lung by a stabilized lipoplex using sodium chloride for complex formation. J. Gene Med. 2005, 7, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.L.; Yu, R.T.; Myers, A.G. Storable arylpalladium(II) reagents for alkene labeling in aqueous media. J. Am. Chem. Soc. 2011, 133, 15870–15873. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belmadi, N.; Berchel, M.; Denis, C.; Berthe, W.; Sibiril, Y.; Le Gall, T.; Haelters, J.-P.; Jaffres, P.-A.; Montier, T. Evaluation of New Fluorescent Lipophosphoramidates for Gene Transfer and Biodistribution Studies after Systemic Administration. Int. J. Mol. Sci. 2015, 16, 26055-26076. https://doi.org/10.3390/ijms161125941
Belmadi N, Berchel M, Denis C, Berthe W, Sibiril Y, Le Gall T, Haelters J-P, Jaffres P-A, Montier T. Evaluation of New Fluorescent Lipophosphoramidates for Gene Transfer and Biodistribution Studies after Systemic Administration. International Journal of Molecular Sciences. 2015; 16(11):26055-26076. https://doi.org/10.3390/ijms161125941
Chicago/Turabian StyleBelmadi, Nawal, Mathieu Berchel, Caroline Denis, Wilfried Berthe, Yann Sibiril, Tony Le Gall, Jean-Pierre Haelters, Paul-Alain Jaffres, and Tristan Montier. 2015. "Evaluation of New Fluorescent Lipophosphoramidates for Gene Transfer and Biodistribution Studies after Systemic Administration" International Journal of Molecular Sciences 16, no. 11: 26055-26076. https://doi.org/10.3390/ijms161125941