
EZH2 in Bladder Cancer, a Promising Therapeutic Target

 , and
, and 
Abstract
:
1. Introduction
2. EZH2 Biological Function
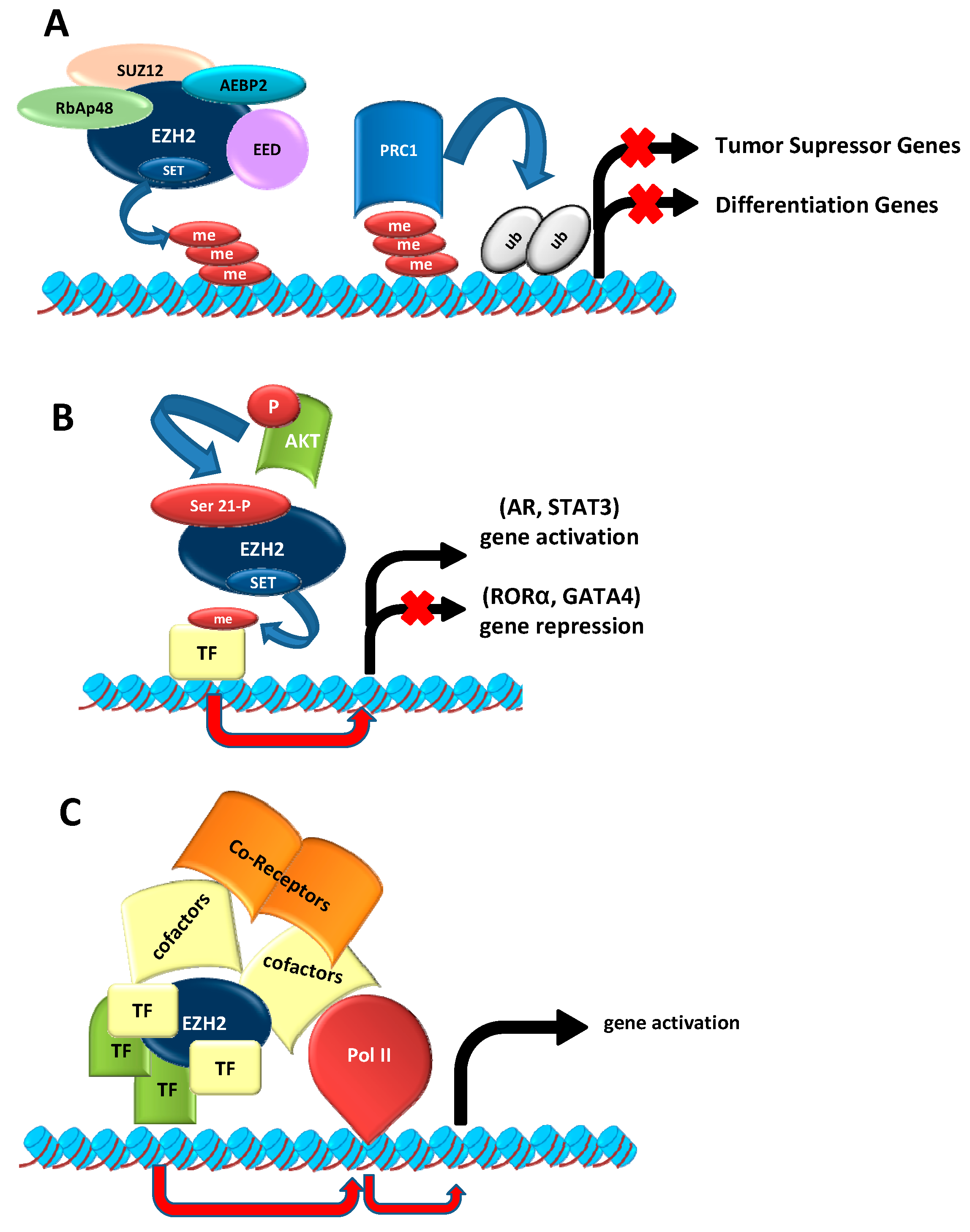
2.1. As Epigenetic Silencer

2.2. EZH2 Non-Canonical Roles
3. Regulation and Crosstalks
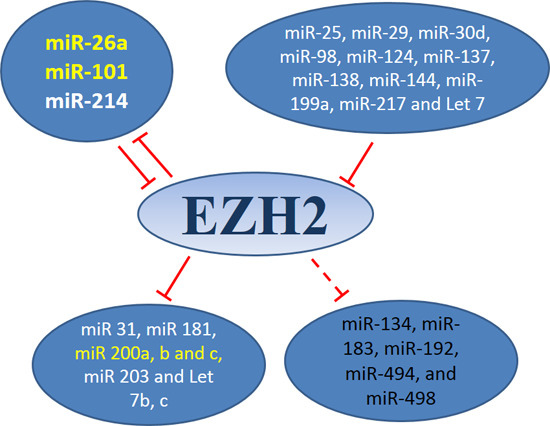
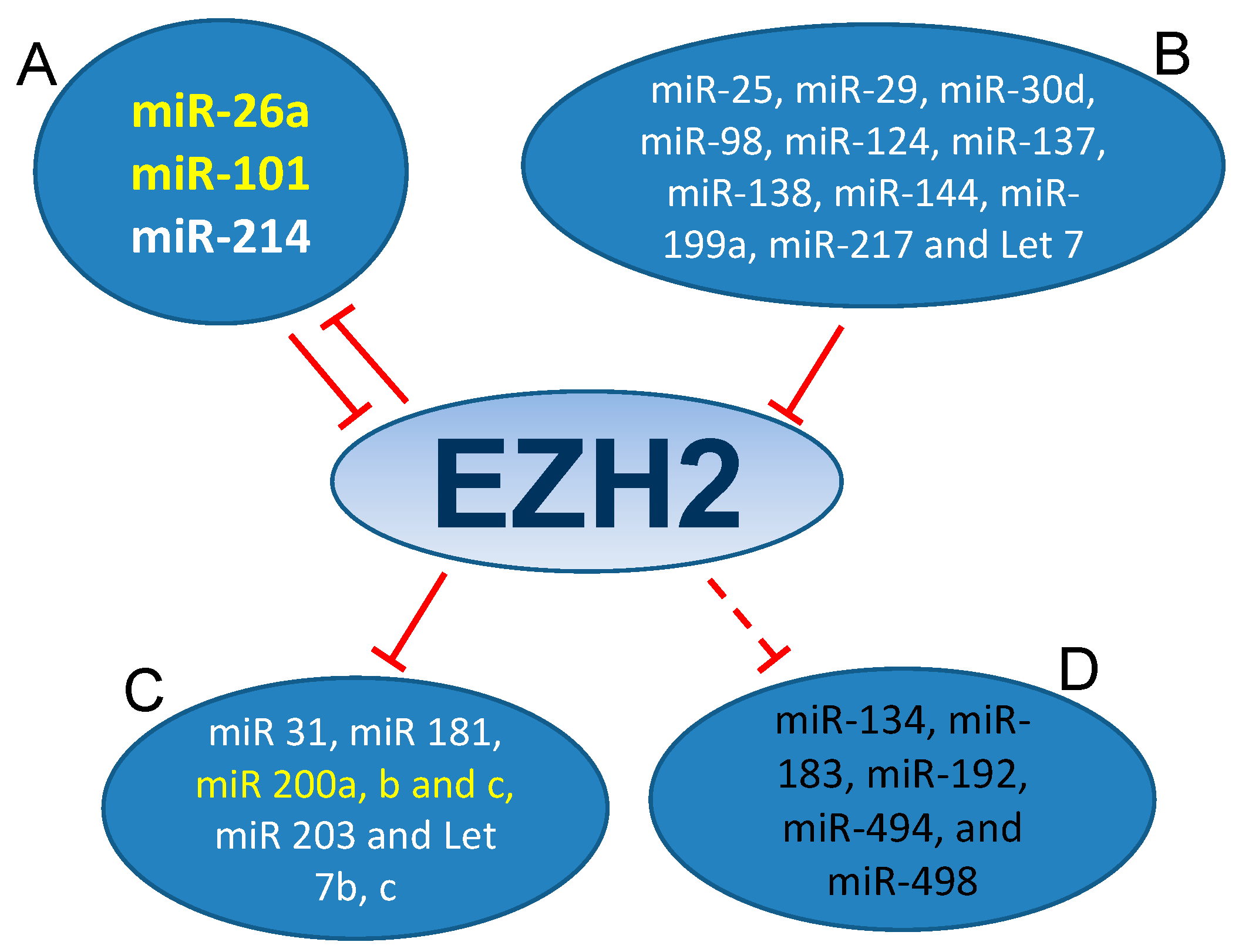
3.1. MiRNAs-EZH2 Interactions

3.2. LncRNAs-EZH2 Interactions
3.3. EZH2 Interaction with Other Epigenetic Enzymes
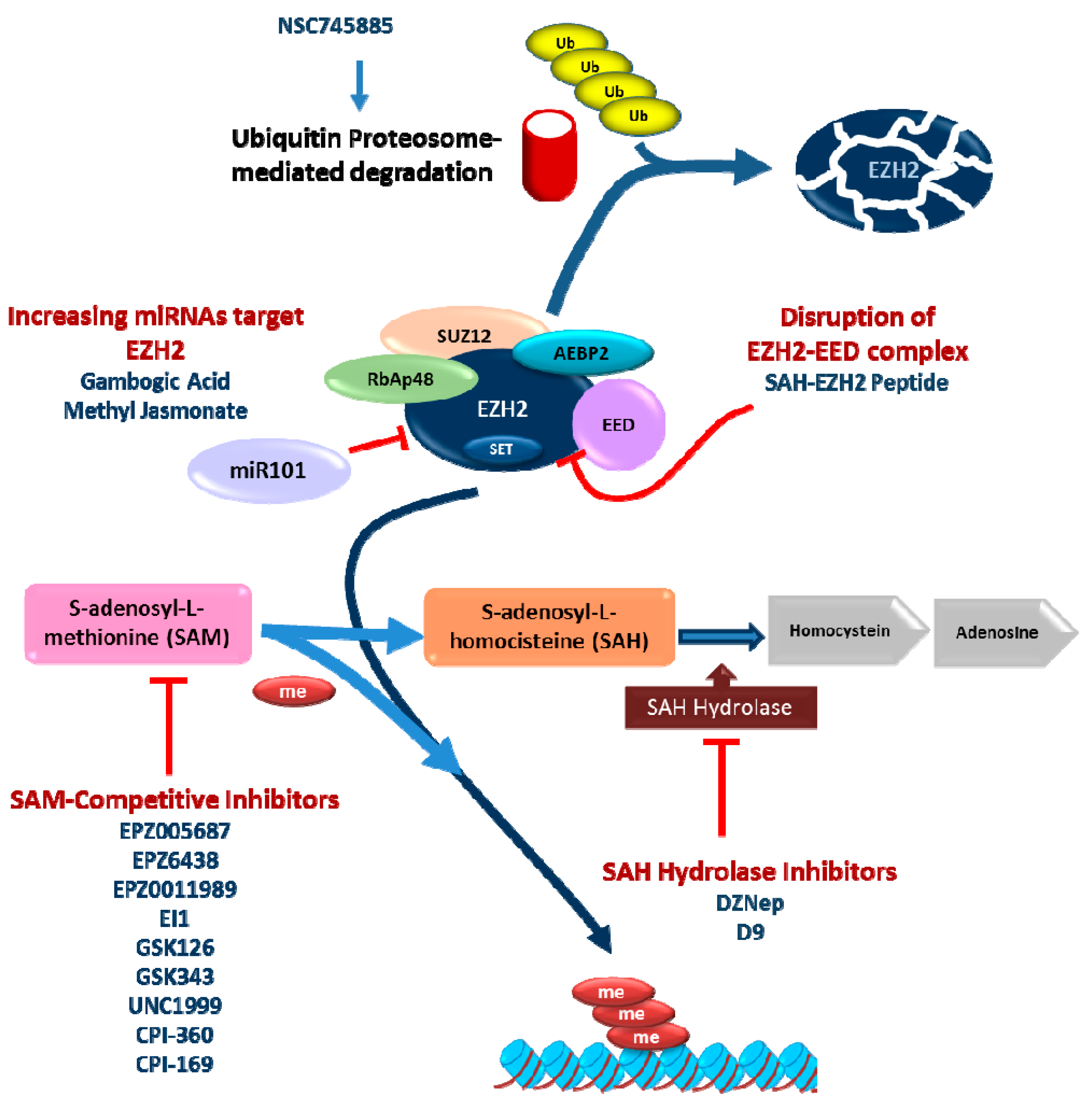
4. Pharmacological Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Mechanism | Specificity to EZH2 | Clinical Status | References |
|---|---|---|---|---|---|
| DZNEP |  | SAH Hydrolase Inhibitor | No | Preclinical | [171,172,173] |
| D9 |  | SAH Hydrolase Inhibitor | No | Preclinical | [174] |
| EPZ005687 |  | SAM-Competitive Inhibitor | Yes | Preclinical | [175] |
| EPZ-6438 |  | SAM-Competitive Inhibitor | Yes | Phase I/II trial | [176] |
| EPZ0011989 |  | SAM-Competitive Inhibitor | Yes | Preclinical | [177] |
| EI1 |  | SAM-Competitive Inhibitor | Yes | Preclinical | [178] |
| GSK126 |  | SAM-Competitive Inhibitor | Yes | Phase I trial | [179] |
| GSK343 |  | SAM-Competitive Inhibitor | Yes | Preclinical | [180,181] |
| UNC1999 |  | SAM-Competitive Inhibitor | Specificity to EZH1/2 | Preclinical | [182,183] |
| CPI-360 |  | SAM-Competitive Inhibitor | Yes | Preclinical | [184] |
| CPI-169 |  | SAM-Competitive Inhibitor | Yes | Preclinical | [184] |
| SAH-EZH2 Peptide | Peptide: FSSNRXKILXRTQILNQEWKQRRIQPV | Disrupts the EZH2-EED complex | Yes | Preclinical | [185] |
| NSC745885 |  | Degradation of EZH2 by proteasome | Yes | Preclinical | [186] |
| GA and MJ |  | Down-regulation of EZH2 expression by mir-101 up-regulation | Yes | Preclinical | [187] |
4.1. S-Adenosyl-l-homocysteine Hydrolase (SAH Hydrolase) Inhibitors

4.2. S-Adenosyl-l-methionine (SAM) Competitive Inhibitors of EZH2
4.3. Other Approaches
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ploeg, M.; Aben, K.K.H.; Kiemeney, L.A. The present and future burden of urinary bladder cancer in the world. World J. Urol. 2009, 27, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Catto, J.W.F.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; la Vecchia, C.; Shariat, S.; et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Abol-Enein, H. Infection: Is it a cause of bladder cancer? Scand. J. Urol. Nephrol. Suppl. 2008, 42, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Blach, O.; Rai, B.; Oates, K.; Franklin, G.; Bramwell, S. An outbreak of schistosomiasis in travellers returning from endemic areas: The importance of rigorous tracing in peer groups exposed to risk of infection. J. Public Health 2012, 34, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Douard, A.; Cornelis, F.; Malvy, D. Urinary schistosomiasis in France. Int. J. Infect. Dis. 2011, 15, e506–e507. [Google Scholar] [CrossRef] [PubMed]
- Oyaert, M.; Lagrange, W.; Smet, G.; de Feyter, K.; Laffut, W. Unexpected urinary Schistosoma infection in a Belgian travel group returning from Malawi. Acta Clin. Belg. 2013, 68, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Mostofi, F.K.; Sobin LH, T.H. Histological Typing of Urinary Bladder Tumours; World Health Organisation: Geva, Switzerland, 1973. [Google Scholar]
- Eble, J.N.; Sauter, G.; Epstein, J.I.; Sesterhenn, I.A.E. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs; IARC Press: Lyon, France, 2004. [Google Scholar]
- Lopez-Beltran, A.; Luque, R.J.; Alvarez-Kindelan, J.; Quintero, A.; Merlo, F.; Requena, M.J.; Montironi, R. Prognostic factors in survival of patients with stage Ta and T1 bladder urothelial tumors: The role of G1-S modulators (p53, p21Waf1, p27Kip1, cyclin D1, and cyclin D3), proliferation index, and clinicopathologic parameters. Am. J. Clin. Pathol. 2004, 122, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2014, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Sauter, G.; Algaba, F.; Amin, M. Tumours of the Urinary System: Non-Invasive Urothelial Neoplasias. In HO Classification of Tumours of the Urinary System and Male Genital Organs; Eble, J.N., Sauter, G., Epstein, J., Sesterhenn, I., Eds.; IARCC Press: Lyon, France, 2004. [Google Scholar]
- NICE (National Institute for Health and Care Excellence). Bladder Cancer: Diagnosis and Management; NICE (National Institute for Health and Care Excellence): London, UK, 2015. [Google Scholar]
- Redelman-Sidi, G.; Glickman, M.S.; Bochner, B.H. The mechanism of action of BCG therapy for bladder cancer—A current perspective. Nat. Rev. Urol. 2014, 11, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.; Sahai, A.; Billia, M.; Dasgupta, P.; Khan, M.S. Recent advances in the diagnosis and treatment of bladder cancer. BMC Med. 2013, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Xylinas, E.; Rink, M.; Margulis, V.; Karakiewicz, P.; Novara, G.; Shariat, S.F. Multifocal carcinoma in situ of the upper tract is associated with high risk of bladder cancer recurrence. Eur. Urol. 2012, 61, 1069–1070. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, R.J.; van der Meijden, A.P.M.; Oosterlinck, W.; Witjes, J.A.; Bouffioux, C.; Denis, L.; Newling, D.W.W.; Kurth, K. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: A combined analysis of 2596 patients from seven EORTC trials. Eur. Urol. 2006, 49, 466–465. [Google Scholar] [PubMed]
- Stenzl, A.; Cowan, N.C.; de Santis, M.; Kuczyk, M.A.; Merseburger, A.S.; Ribal, M.J.; Sherif, A.; Witjes, J.A. Treatment of muscle-invasive and metastatic bladder cancer: Update of the EAU guidelines. Eur. Urol. 2011, 59, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Karl, A.; Carroll, P.R.; Gschwend, J.E.; Knüchel, R.; Montorsi, F.; Stief, C.G.; Studer, U.E. The impact of lymphadenectomy and lymph node metastasis on the outcomes of radical cystectomy for bladder cancer. Eur. Urol. 2009, 55, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Hurst, C.D.; Taylor, C.F.; Gregory, W.M.; Harnden, P.; Knowles, M.A. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin. Cancer Res. 2009, 15, 6008–6017. [Google Scholar] [CrossRef] [PubMed]
- Kompier, L.C.; Lurkin, I.; van der Aa, M.N.M.; van Rhijn, B.W.G.; van der Kwast, T.H.; Zwarthoff, E.C. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS ONE 2010, 5, e13821. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, M.; Martínez-Fernández, M.; García-Escudero, R.; Villacampa, F.; Marqués, M.; Saiz-Ladera, C.; Duarte, J.; Martínez, V.; Gómez, M.J.; Martín, M.L.; et al. PIK3CA gene alterations in bladder cancer are frequent and associate with reduced recurrence in non-muscle invasive tumors. Mol. Carcinog. 2013. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Al-Rohil, R.N.; Nazeer, T.; Sheehan, C.E.; Otto, G.A.; He, J.; Palmer, G.; Yelensky, R.; Lipson, D.; et al. Advanced urothelial carcinoma: Next-generation sequencing reveals diverse genomic alterations and targets of therapy. Mod. Pathol. 2014, 27, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Teo, M.T.W.; Dyrskjøt, L.; Nsengimana, J.; Buchwald, C.; Snowden, H.; Morgan, J.; Jensen, J.B.; Knowles, M.A.; Taylor, G.; Barrett, J. H.; et al. Next-generation sequencing identifies germline MRE11A variants as markers of radiotherapy outcomes in muscle-invasive bladder cancer. Ann. Oncol. 2014, 25, 877–883. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Available online: https://tcga-data.nci.nih.gov/tcga/ (accessed on 5 November 2015).
- Damrauer, J.S.; Hoadley, K.A.; Chism, D.D.; Fan, C.; Tiganelli, C.J.; Wobker, S.E.; Yeh, J.J.; Milowsky, M.I.; Iyer, G.; Parker, J.S.; et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc. Natl. Acad. Sci. USA 2014, 111, 3110–3115. [Google Scholar] [CrossRef] [PubMed]
- Sjödahl, G.; Lauss, M.; Lövgren, K.; Chebil, G.; Gudjonsson, S.; Veerla, S.; Patschan, O.; Aine, M.; Fernö, M.; Ringnér, M.; et al. A molecular taxonomy for urothelial carcinoma. Clin. Cancer Res. 2012, 18, 3377–3386. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.-L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Aine, M.; Eriksson, P.; Liedberg, F.; Sjödahl, G.; Höglund, M. Biological determinants of bladder cancer gene expression subtypes. Sci. Rep. 2015, 5, 10957. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, M.; Varambally, S.; Cao, Q.; Chinnaiyan, A.M.; Ferguson, D.O.; Merajver, S.D.; Kleer, C.G. The Polycomb group protein EZH2 impairs DNA repair in breast epithelial cells. Neoplasia 2005, 7, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Yu, J. EZH2, an epigenetic driver of prostate cancer. Protein Cel. 2013, 4, 331–41. [Google Scholar] [CrossRef] [PubMed]
- Grzenda, A.; Ordog, T.; Urrutia, R. Polycomb and the emerging epigenetics of pancreatic cancer. J. Gastrointest. Cancer 2011, 42, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Rauch, T.A. DNA methylation patterns in lung carcinomas. Semin. Cancer Biol. 2009, 19, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Deb, G.; Singh, A.K.; Gupta, S. EZH2: Not EZHY (easy) to deal. Mol. Cancer Res. 2014, 12, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hung, M.-C. Regulation and Role of EZH2 in Cancer. Cancer Res. Treat. 2014, 46, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Hurt, E.M.; Mathews, L.A.; Cabarcas, S.M.; Sun, L.; Marquez, V.E.; Danesi, R.; Farrar, W.L. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol. Cancer 2011, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Pietersen, A.M.; Horlings, H.M.; Hauptmann, M.; Langerød, A.; Ajouaou, A.; Cornelissen-Steijger, P.; Wessels, L.F.; Jonkers, J.; van de Vijver, M.J.; van Lohuizen, M. EZH2 and BMI1 inversely correlate with prognosis and TP53 mutation in breast cancer. Breast Cancer Res. 2008, 10, R109. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, C.; Lander, G.C.; Maiolica, A.; Herzog, F.; Aebersold, R.; Nogales, E. Molecular architecture of human polycomb repressive complex 2. Elife 2012, 1, e00005. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shen, L.; Cheng, A. S.; Ahmed, S.; Boumber, Y.; Charo, C.; Yamochi, T.; Urano, T.; Furukawa, K.; Kwabi-Addo, B.; et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat. Genet. 2008, 40, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, M.; Sauvageau, G. Polycomb group proteins: Multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 2010, 7, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Grzenda, A.; Lomberk, G.; Svingen, P.; Mathison, A.; Calvo, E.; Iovanna, J.; Xiong, Y.; Faubion, W.; Urrutia, R. Functional characterization of EZH2β reveals the increased complexity of EZH2 isoforms involved in the regulation of mammalian gene expression. Epigenet. Chromatin 2013, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asangani, I.A.; Ateeq, B.; Cao, Q.; Dodson, L.; Pandhi, M.; Kunju, L.P.; Mehra, R.; Lonigro, R.J.; Siddiqui, J.; Palanisamy, N.; et al. Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol. Cell. 2013, 49, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Mani, R.-S.; Ateeq, B.; Dhanasekaran, S.M.; Asangani, I.A.; Prensner, J.R.; Kim, J.H.; Brenner, J.C.; Jing, X.; Cao, X.; et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell 2011, 20, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Rao, M.; Humphries, A.E.; Hong, J.A.; Liu, F.; Yang, M.; Caragacianu, D.; Schrump, D.S. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009, 69, 3570–3578. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-J.; Yang, J.-Y.; Xia, W.; Chen, C.-T.; Xie, X.; Chao, C.-H.; Woodward, W.A.; Hsu, J.-M.; Hortobagyi, G.N.; Hung, M.-C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.E.; Li, X.; Toy, K.; DuPrie, M.; Ventura, A.C.; Banerjee, M.; Ljungman, M.; Merajver, S.D.; Kleer, C.G. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene 2009, 28, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Fukamachi, K.; Tsuda, H.; Ito, K.; Ito, Y.; Ochiai, A. RAS oncogenic signal upregulates EZH2 in pancreatic cancer. Biochem. Biophys. Res. Commun. 2012, 417, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Zaslavsky, A.; Fedele, G.; McLaughlin, S.K.; Reczek, E.E.; de Raedt, T.; Guney, I.; Strochlic, D.E.; Macconaill, L.E.; Beroukhim, R.; et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 2010, 16, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.-L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.-C.; Chen, C.-T.; Ping, B.; Otte, A.P.; Hung, M.-C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.-H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y. T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, J.S.; Kim, H.; Kim, K.; Park, H.; Kim, J.-Y.; Lee, S.H.; Kim, I.S.; Kim, J.; Lee, M.; et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 2012, 48, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.M.; Montagnani Marelli, M.; Motta, M.; Limonta, P. Role of the orphan nuclear receptor RORα in the control of the metastatic behavior of androgen-independent prostate cancer cells. Oncol. Rep. 2002, 9, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Kim, I.S.; Kim, H.; Lee, J.S.; Kim, K.; Yim, H.Y.; Jeong, J.; Kim, J.H.; Kim, J.-Y.; Lee, H.; et al. RORα attenuates Wnt/β-catenin signaling by PKCα-dependent phosphorylation in colon cancer. Mol. Cell 2010, 37, 183–195. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Shen, X.; Ma, Q.; Cao, J.; von Gise, A.; Zhou, P.; Wang, G.; Marquez, V.E.; Orkin, S.H.; Pu, W.T. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012, 26, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yan, Y.; Wang, X.; Jiang, Y.; Xu, H.E. EZH2: Biology, disease, and structure-based drug discovery. Acta Pharmacol. Sin. 2014, 35, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Liang, J.; Yang, X.; Wang, Y.; Zhao, Y.; Wu, H.; Sun, L.; Zhang, Y.; Chen, Y.; Li, R.; et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol. Cell. Biol. 2007, 27, 5105–5119. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.K.M.; Liou, Y.C.; Yu, Q. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.-Y.; Jun, S.; Lee, M.; Kim, H.-C.; Wang, X.; Ji, H.; McCrea, P.D.; Park, J.-I. PAF and EZH2 induce Wnt/β-catenin signaling hyperactivation. Mol. Cell 2013, 52, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Ng, S.-B.; Tay, J.L.-S.; Lin, B.; Koh, T.L.; Tan, J.; Selvarajan, V.; Liu, S.-C.; Bi, C.; Wang, S.; et al. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood 2013, 121, 4512–4520. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.P.; Pagliarulo, V.; Yang, D.; Waldman, F.M.; Datar, R.H.; Skinner, D.G.; Groshen, S.; Cote, R.J. Generation of a Concise gene panel for outcome prediction in urinary bladder cancer. J. Clin. Oncol. 2009, 27, 3929–3937. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J.; Chang, H. Y.; van de Rijn, M.; et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.L.; Lay, E.J.; Jian, W.; Parra, D.; Chan, K.S. STAT3 activation in urothelial stem cells leads to direct progression to invasive bladder cancer. Cancer Res. 2012, 72, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ying, L.; Tian, Y.; Yang, P.; Zhu, Y.; Wang, Z.; Qiu, F.; Lin, J. miR-144 downregulation increases bladder cancer cell proliferation by targeting EZH2 and regulating Wnt signaling. FEBS J. 2013, 280, 4531–4538. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplied in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Iwata, T.; Zheng, Q.; Bethel, C.; Yegnasubramanian, S.; de Marzo, A.M. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget 2011, 2, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Zippo, A.; Krepelova, A.; Cherubini, A.; Rocchigiani, M. Oliviero, S. Myc regulates the transcription of the PRC2 gene to control the expression of developmental genes in embryonic stem cells. Mol. Cell. Biol. 2012, 32, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Milyavsky, M.; Shats, I.; Erez, N.; Goldfinger, N. Rotter, V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene 2004, 23, 5759–5769. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Voulgaris, E.; Vartholomatos, G.; Hatzimichael, E. Non-coding RNAs and EZH2 interactions in cancer: Long and short tales from the transcriptome. Int. J. Cancer 2013, 133, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Zhang, Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J. Biol. Chem. 2011, 286, 28511–28519. [Google Scholar] [CrossRef] [PubMed]
- Riising, E.M.; Boggio, R.; Chiocca, S.; Helin, K. Pasini, D. The polycomb repressive complex 2 is a potential target of SUMO modifications. PLoS ONE 2008, 3, e2704. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Cole, M.D. MYC Acts via the PTEN Tumor Suppressor to Elicit Autoregulation and Genome-Wide Gene Repression by Activation of the Ezh2 Methyltransferase. Cancer Res. 2012, 73, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, X.; Jia, L.-T.; Hu, S.-J.; Zhao, J.; Yang, J.-D.; Wen, W.-H.; Wang, Z.; Wang, T.; Zhao, J.; et al. c-Myc-mediated epigenetic silencing of MicroRNA-101 contributes to dysregulation of multiple pathways in hepatocellular carcinoma. Hepatology 2014, 59, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Bullinger, L.; Klapproth, K.; Fiedler, K.; Kestler, H.A.; Barth, T.F.E.; Möller, P.; Stilgenbauer, S.; Pollack, J.R.; Wirth, T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 2008, 112, 4202–4212. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.-Z.; Li, C.-X.; Zhang, Y.; Weng, M.-Z.; Zhang, M.; Qin, Y.-Y.; Gong, W.; Quan, Z.-W. Long non-coding RNA HOTAIR, a c-Myc activated driver of malignancy, negatively regulates miRNA-130a in gallbladder cancer. Mol. Cancer 2014, 13, 156. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Li, G.; Son, J.; Xu, C.-F.; Margueron, R.; Neubert, T.A.; Reinberg, D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010, 24, 2615–2620. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Esteller, M. DNA methylomes, histone codes and miRNAs: Tying it all together. Int. J. Biochem. Cell. Biol. 2009, 41, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Iliopoulos, D. Epigenetic aberrations during oncogenesis. Cell. Mol. Life Sci. 2011, 68, 1681–1702. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Mancini-Dinardo, D.; Steele, S.J.S.; Levorse, J.M.; Ingram, R.S.; Tilghman, S.M. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006, 20, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Cao, Q.; Mani, R.-S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Juan, A.H.; Kumar, R.M.; Marx, J.G.; Young, R.A.; Sartorelli, V. Mir-214-dependent regulation of the polycomb protein EZH2 in skeletal muscle and embryonic stem cells. Mol. Cell 2009, 36, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Guo, J.-F.; Liu, D.-L.; Liu, Q.; Wang, J.-J. MicroRNA-101 exerts tumor-suppressive functions in non-small cell lung cancer through directly targeting enhancer of zeste homolog 2. J. Thorac. Oncol. 2011, 6, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Tornincasa, M.; Pallante, P.; Federico, A.; Borbone, E.; Pierantoni, G.M.; Fusco, A. Down-regulation of the miR-25 and miR-30d contributes to the development of anaplastic thyroid carcinoma targeting the polycomb protein EZH2. J. Clin. Endocrinol. Metab. 2012, 97, E710–E718. [Google Scholar] [CrossRef] [PubMed]
- Tsukigi, M.; Bilim, V.; Yuuki, K.; Ugolkov, A.; Naito, S.; Nagaoka, A.; Kato, T.; Motoyama, T.; Tomita, Y. Re-expression of miR-199a suppresses renal cancer cell proliferation and survival by targeting GSK-3β. Cancer Lett. 2012, 315, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Heath, E.; Chen, W.; Cher, M. L.; Powell, I.; Heilbrun, L.; Li, Y.; Ali, S.; Sethi, S.; Hassan, O.; et al. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS ONE 2012, 7, e33729. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, B.; Wapinski, O.L.; Tsai, M.-C.; Qu, K.; Zhang, J.; Carlson, J. C.; Lin, M.; Fang, F.; Gupta, R.A.; et al. Targeted disruption of HOTAIR leads to homeotic transformation and gene derepression. Cell. Rep. 2013, 5, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, H.; Zhao, M.; Lv, Z.; Zhang, X.; Qin, X.; Wang, H.; Wang, S.; Su, J.; Lv, X.; et al. MiR-138 inhibits tumor growth through repression of EZH2 in non-small cell lung cancer. Cell. Physiol. Biochem. 2013, 31, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-D.; Yuan, Y.; Zhuang, C.-W.; Li, B.-L.; Gong, D.-J.; Wang, S.-G.; Zeng, Z.-Y.; Cheng, H.-Z. MicroRNA-98 and microRNA-214 post-transcriptionally regulate enhancer of zeste homolog 2 and inhibit migration and invasion in human esophageal squamous cell carcinoma. Mol. Cancer 2012, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-J.; Ruan, H.-J.; He, X.-J.; Ma, Y.-Y.; Jiang, X.-T.; Xia, Y.-J.; Ye, Z.-Y.; Tao, H.-Q. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur. J. Cancer 2010, 46, 2295–303. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, O.; Takamori, H.; Iwatsuki, M.; Baba, Y.; Sakamoto, Y.; Tanaka, H.; Chikamoto, A.; Horino, K.; Beppu, T.; Kanemitsu, K.; et al. Carcinogenesis of intraductal papillary mucinous neoplasm of the pancreas: Loss of microRNA-101 promotes overexpression of histone methyltransferase EZH2. Ann. Surg. Oncol. 2012, 19 (Suppl. 3), S565–S571. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Liang, G.; Liu, C.-C.; Wolff, E.M.; Tsai, Y.C.; Ye, W.; Zhou, X.; Jones, P.A. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res. 2009, 69, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Tzatsos, A.; Paskaleva, P.; Lymperi, S.; Contino, G.; Stoykova, S.; Chen, Z.; Wong, K.-K.; Bardeesy, N. Lysine-specific demethylase 2B (KDM2B)-let-7-enhancer of zester homolog 2 (EZH2) pathway regulates cell cycle progression and senescence in primary cells. J. Biol. Chem. 2011, 286, 33061–33069. [Google Scholar] [CrossRef] [PubMed]
- Kottakis, F.; Polytarchou, C.; Foltopoulou, P.; Sanidas, I.; Kampranis, S.C.; Tsichlis, P.N. FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol. Cell 2011, 43, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Padi, S.K.R.; Tindall, D.J.; Guo, B. Polycomb protein EZH2 suppresses apoptosis by silencing the proapoptotic miR-31. Cell. Death Dis. 2014, 5, e1486. [Google Scholar] [CrossRef] [PubMed]
- Au, S.L.-K.; Wong, C.C.-L.; Lee, J. M.-F.; Fan, D.N.-Y.; Tsang, F.H.; Ng, I.O.-L.; Wong, C.-M. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 2012, 56, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D.; et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011, 29, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qiu, M.; Tan, G.; Liang, Z.; Qin, Y.; Chen, L.; Chen, H.; Liu, J. miR-200c inhibits invasion, migration and proliferation of bladder cancer cells through down-regulation of BMI-1 and E2F3. J. Transl. Med. 2014, 12, 305. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Martínez-Fernández, M.; Dueñas, M.; García-Escudero, R.; Alfaya, B.; Villacampa, F.; Saiz-Ladera, C.; Costa, C.; Oteo, M.; Duarte, J.; et al. In vivo disruption of an Rb-E2F-EZH2 signaling loop causes bladder cancer. Cancer Res. 2014, 74, 6565–6577. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, E.D.; Bramsen, J.B.; Hulf, T.; Dyrskjøt, L.; Ramanathan, R.; Hansen, T.B.; Villadsen, S.B.; Gao, S.; Ostenfeld, M.S.; Borre, M.; et al. Coordinated epigenetic repression of the miR-200 family and miR-205 in invasive bladder cancer. Int. J. Cancer 2011, 128, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, M.; Dueñas, M.; Feber, A.; Segovia, C.; García-Escudero, R.; Rubio, C.; López-Calderón, F.F.; Díaz-García, C.; Villacampa, F.; Duarte, J.; et al. A Polycomb-miR200 loop regulates clinical outcome in bladder cancer. Oncotarget 2015, in press. [Google Scholar]
- Wang, J.; Zhang, X.; Wang, L.; Dong, Z.; Du, L.; Yang, Y.; Guo, Y.; Wang, C. Downregulation of urinary cell-free microRNA-214 as a diagnostic and prognostic biomarker in bladder cancer. J. Surg. Oncol. 2015, 111, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhang, Z.; Tan, Z.; He, R.; Zeng, X.; Xie, Y.; Li, S.; Tang, G.; Tang, H.; He, X. MicroRNA-124 inhibits proliferation and induces apoptosis by directly repressing EZH2 in gastric cancer. Mol. Cell. Biochem. 2014, 392, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Neo, W.H.; Yap, K.; Lee, S.H.; Looi, L.S.; Khandelia, P.; Neo, S.X.; Makeyev, E.V.; Su, I. MicroRNA miR-124 controls the choice between neuronal and astrocyte differentiation by fine-tuning EZH2 expression. J. Biol. Chem. 2014, 289, 20788–20801. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Liao, Y.-J.; Cai, M.-Y.; Liu, Y.-H.; Liu, T.-H.; Chen, S.-P.; Bian, X.-W.; Guan, X.-Y.; Lin, M.C.; Zeng, Y.-X.; et al. The putative tumour suppressor microRNA-124 modulates hepatocellular carcinoma cell aggressiveness by repressing ROCK2 and EZH2. Gut 2012, 61, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Xiu, Y.; Liu, Z.; Xia, S.; Jin, C.; Yin, H.; Zhao, W.; Wu, Q. MicroRNA-137 upregulation increases bladder cancer cell proliferation and invasion by targeting PAQR3. PLoS ONE 2014, 9, e109734. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, S.; Lin, Y.; Chen, H.; Hu, Z.; Mao, Y.; Xu, X.; Wu, J.; Zhu, Y.; Zheng, X.; et al. MicroRNA-124-3p inhibits cell migration and invasion in bladder cancer cells by targeting ROCK1. J. Transl. Med. 2013, 11, 276. [Google Scholar] [CrossRef] [PubMed]
- Kunej, T.; Godnic, I.; Ferdin, J.; Horvat, S.; Dovc, P.; Calin, G.A. Epigenetic regulation of microRNAs in cancer: An integrated review of literature. Mutat. Res. Mol. Mech. Mutagen. 2011, 717, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, S.P.; Koch, C.M.; Clelland, G.K.; Willcox, S.; Fowler, J.C.; Stewart, R.; Lako, M.; Dunham, I.; Armstrong, L. Epigenetic marking prepares the human HOXA cluster for activation during differentiation of pluripotent cells. Stem Cells 2008, 26, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Fraser, P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular signals of epigenetic states. Science 2010, 330, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Li, Z.; Wang, W.; Zeng, Y.; Liu, Z.; Qiu, J. Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett. 2013, 333, 213–221. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Cai, Q.; Sun, F.; Zhong, G.; Wang, P.; Liu, H.; Luo, J.; Yu, H.; Huang, J.; Lin, T. linc-UBC1 physically associates with polycomb repressive complex 2 (PRC2) and acts as a negative prognostic factor for lymph node metastasis and survival in bladder cancer. Biochim. Biophys. Acta 2013, 1832, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Tano, K.; Akimitsu, N. Long non-coding RNAs in cancer progression. Front. Genet. 2012, 3, 219. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, M.; Feber, A.; Dueñas, M.; Segovia, C.; Rubio, C.; Fernandez, M.; Villacampa, F.; Duarte, J.; Lopez-Calderon, F.F.; Gomez-Rodriguez, M.J.; et al. Analysis of the Polycomb-related lncRNAs HOTAIR and ANRIL in bladder cancer. Clin. Epigenet. 2015, in press. [Google Scholar]
- Geng, Y.J.; Xie, S.L.; Li, Q.; Ma, J.; Wang, G.Y. Large intervening non-coding RNA HOTAIR is associated with hepatocellular carcinoma progression. J. Int. Med. Res. 2011, 39, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Yu, J.; Dong, R.; Qiu, H. A high level of circulating HOTAIR is associated with progression and poor prognosis of cervical cancer. Tumour Biol. 2014, 36, 1661–1665. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Nguyen, H.T.; Burow, M.E.; Zhuo, Y.; El-Dahr, S.S.; Yao, X.; Cao, S.; Flemington, E.K.; Nephew, K.P.; Fang, F.; et al. Elevated expression of long intergenic non-coding RNA HOTAIR in a basal-like variant of MCF-7 breast cancer cells. Mol. Carcinog. 2014, 54, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Schorderet, P.; Duboule, D. Structural and functional differences in the long non-coding RNA HOTAIR in mouse and human. PLoS Genet. 2011, 7, e1002071. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Shin, Y.K. Epigenetic regulation of cancer-associated genes in ovarian cancer. Int. J. Mol. Sci. 2011, 12, 983–1008. [Google Scholar] [CrossRef] [PubMed]
- Gal-Yam, E.N.; Egger, G.; Iniguez, L.; Holster, H.; Einarsson, S.; Zhang, X.; Lin, J.C.; Liang, G.; Jones, P.A.; Tanay, A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. USA 2008, 105, 12979–12984. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Shi, Z.; Liu, X.; Zhang, A.; Han, L.; Jiang, K.; Kang, C.; Zhang, Q. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett. 2015, 359, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; LeRoy, G.; Drury, W.J.; Zee, B.M.; Son, J.; Beck, D.B.; Young, N.L.; Garcia, B.A.; Reinberg, D. Asymmetrically modified nucleosomes. Cell 2012, 151, 181–193. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; Chepelev, I.; DiMaggio, P.A.; Blanco, M.A.; Zee, B.M.; Zhao, K.; Garcia, B.A. Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol. 2012, 13, R68. [Google Scholar] [CrossRef] [PubMed]
- Escamilla-Del-Arenal, M.; da Rocha, S.T.; Spruijt, C.G.; Masui, O.; Renaud, O.; Smits, A.H.; Margueron, R.; Vermeulen, M.; Heard, E. Cdyl, a new partner of the inactive X chromosome and potential reader of H3K27me3 and H3K9me2. Mol. Cell. Biol. 2013, 33, 5005–5020. [Google Scholar] [CrossRef] [PubMed]
- Boros, J.; Arnoult, N.; Stroobant, V.; Collet, J.-F.; Decottignies, A. Polycomb repressive complex 2 and H3K27me3 cooperate with H3K9 methylation to maintain heterochromatin protein 1α at chromatin. Mol. Cell. Biol. 2014, 34, 3662–3674. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Pontis, J.; Fritsch, L.; Robin, P.; Portoso, M.; Proux, C.; Margueron, R.; Ait-Si-Ali, S. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell 2014, 53, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.R.; Bahirvani, A.G.; Rao, V.K.; Bharathy, N.; Ow, J.R.; Taneja, R. G9a, a multipotent regulator of gene expression. Epigenetics 2013, 8, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Roger, T.; Lugrin, J.; Le Roy, D.; Goy, G.; Mombelli, M.; Koessler, T.; Ding, X.C.; Chanson, A.-L.; Reymond, M.K.; Miconnet, I.; et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011, 117, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Niegisch, G.; Knievel, J.; Koch, A.; Hader, C.; Fischer, U.; Albers, P.; Schulz, W.A. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol. 2013, 31, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Hoffmann, M.J.; Koch, A.; Ulrich, S.M.; Schulz, W.A.; Niegisch, G. Histone deacetylase 8 is deregulated in urothelial cancer but not a target for efficient treatment. J. Exp. Clin. Cancer Res. 2014, 33, 59. [Google Scholar] [CrossRef] [PubMed]
- Poyet, C.; Jentsch, B.; Hermanns, T.; Schweckendiek, D.; Seifert, H.-H.; Schmidtpeter, M.; Sulser, T.; Moch, H.; Wild, P.J.; Kristiansen, G. Expression of histone deacetylases 1, 2 and 3 in urothelial bladder cancer. BMC Clin. Pathol. 2014, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.A.B.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Varier, R.A.; Timmers, H.T.M. Histone lysine methylation and demethylation pathways in cancer. Biochim. Biophys. Acta 2011, 1815, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Rotili, D.; Mai, A. Targeting histone demethylases: A new avenue for the fight against cancer. Genes Cancer 2011, 2, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- Hong, S.; Cho, Y.-W.; Yu, L.-R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Bayliss, P.E.; Rinn, J.L.; Whetstine, J.R.; Wang, J.K.; Chen, S.; Iwase, S.; Alpatov, R.; Issaeva, I.; Canaani, E.; et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 2007, 449, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.-P.; Reinberg, D.; di Croce, L.; Shiekhattar, R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, S.; Liu, H.-M.; Zhang, Y.-B.; Blair, C.A.; Mercola, D.; Sassone-Corsi, P.; Zi, X. Histone lysine-specific methyltransferases and demethylases in carcinogenesis: New targets for cancer therapy and prevention. Curr. Cancer Drug Targets 2013, 13, 558–579. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Dancik, G.M.; Im, K.M.; Edwards, M.G.; Turan, S.; Brown, J.; Ruiz-Rodriguez, C.; Owens, C.; Costello, J.C.; Guo, G.; et al. Concurrent alterations in TERT, KDM6A, and the BRCA pathway in bladder cancer. Clin. Cancer Res. 2014, 20, 4935–4948. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; Welstead, G.G.; Trimarchi, T.; Bakogianni, S.; Xu, L.; Loizou, E.; Holmfeldt, L.; Strikoudis, A.; King, B.; et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014, 514, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.; Helin, K. Histone methyltransferases in cancer. Semin. Cell. Dev. Biol. 2010, 21, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Baugé, C.; Bazille, C.; Girard, N.; Lhuissier, E.; Boumediene, K. Histone methylases as novel drug targets: Developing inhibitors of EZH2. Future Med. Chem. 2014, 6, 1943–1965. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.M.; Tan, P.B.O.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bi, C.; Cheong, L.L.; Liu, S.C.; Huang, G.; Zhou, J.; Yu, Q.; Chen, C.-S.; Chng, W.J. Determinants of sensitivity to DZNep induced apoptosis in multiple myeloma cells. PLoS ONE 2011, 6, e21583. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lim, C.Z.H.; Li, Z.; Lee, P.L.; Yatim, S.M.J.M.; Guan, P.; Li, J.; Zhou, J.; Pan, J.; Chng, W.-J.; et al. Functional characterization of D9, a novel deazaneplanocin A (DZNep) analog, in targeting acute myeloid leukemia (AML). PLoS ONE 2015, 10, e0122983. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Kuntz, K.W.; Knutson, S.K.; Warholic, N.M.; Keilhack, H.; Wigle, T.J.; Raimondi, A.; Klaus, C.R.; Rioux, N.; Yokoi, A.; et al. EPZ011989, a potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS Med. Chem. Lett. 2015, 6, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Chan, H.; Teng, L.; Li, L.; Chuai, S.; Zhang, R.; Zeng, J.; Li, M.; Fan, H.; Lin, Y.; et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 21360–21365. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; van Aller, G.S.; Liu, Y.; Graves, A.P.; Iii, A.D.P.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Tian, X.; LaFrance, L.V.; Duquenne, C.; Suarez, D.P.; Newlander, K.A.; Romeril, S.P.; Burgess, J.L.; Grant, S.W.; Brackley, J.A.; et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med. Chem. Lett. 2012, 3, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Garipov, A.; Li, H.; Conejo-Garcia, J.R.; Speicher, D.W.; Zhang, R. Three-dimensional culture sensitizes epithelial ovarian cancer cells to EZH2 methyltransferase inhibition. Cell Cycle 2013, 12, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; On, D.M.; Ma, A.; Parton, T.; Konze, K.D.; Pattenden, S.G.; Allison, D.F.; Cai, L.; Rockowitz, S.; Liu, S.; et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 2015, 125, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; Macnevin, C.J.; Liu, F.; Gao, C.; Huang, X.-P.; Kuznetsova, E.; et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Bradley, W.D.; Arora, S.; Busby, J.; Balasubramanian, S.; Gehling, V.S.; Nasveschuk, C.G.; Vaswani, R.G.; Yuan, C.-C.; Hatton, C.; Zhao, F.; et al. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem. Biol. 2014, 21, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bird, G.H.; Neff, T.; Guo, G.; Kerenyi, M.A.; Walensky, L.D.; Orkin, S.H. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol. 2013, 9, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Huang, H.; Wu, H.; Tsai, Y.; Chuang, M. Pharmacologic down-regulation of EZH2 suppresses bladder cancer in vitro and in vivo. Oncotarget 2014, 5, 10342–10355. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiang, W.; Wang, M.; Huang, T.; Xiao, X.; Wang, L.; Tao, D.; Dong, L.; Zeng, F.; Jiang, G. Methyl jasmonate sensitizes human bladder cancer cells to gambogic acid-induced apoptosis through down-regulation of EZH2 expression by miR-101. Br. J. Pharmacol. 2014, 171, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A.; Solomon, M.E.; Richon, V.M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 2009, 8, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Helin, K.; Dhanak, D. Chromatin proteins and modifications as drug targets. Nature 2013, 502, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Balasubramanian, S.; Chew, Y.C.; Han, B.; Marquez, V.E.; Eckert, R.L. (−)-Epigallocatechin-3-gallate and DZNep reduce polycomb protein level via a proteasome-dependent mechanism in skin cancer cells. Carcinogenesis 2011, 32, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Takashina, T.; Kinoshita, I.; Kikuchi, E.; Shimizu, Y.; Sakakibara-Konishi, J.; Oizumi, S.; Marquez, V.E.; Nishimura, M.; Dosaka-Akita, H. Epigenetic therapy with 3-deazaneplanocin A, an inhibitor of the histone methyltransferase EZH2, inhibits growth of non-small cell lung cancer cells. Lung Cancer 2012, 78, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Hibino, S.; Saito, Y.; Muramatsu, T.; Otani, A.; Kasai, Y.; Kimura, M.; Saito, H. Inhibitors of enhancer of zeste homolog 2 (EZH2) activate tumor-suppressor microRNAs in human cancer cells. Oncogenesis 2014, 3, e104. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.L.; Itahana, Y.; Lei, Z.D.; Chia, N.-Y.; Wu, Y.; Yu, Y.; Zhang, S.L.; Thike, A.A.; Pandey, A.; Rozen, S.; et al. TP53 genomic status regulates sensitivity of gastric cancer cells to the histone methylation inhibitor 3-deazaneplanocin A (DZNep). Clin. Cancer Res. 2012, 18, 4201–4212. [Google Scholar] [CrossRef] [PubMed]
- Braun, F.K.; Mathur, R.; Sehgal, L.; Wilkie-Grantham, R.; Chandra, J.; Berkova, Z.; Samaniego, F. Inhibition of methyltransferases accelerates degradation of cFLIP and sensitizes B-cell lymphoma cells to TRAIL-induced apoptosis. PLoS ONE 2015, 10, e0117994. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y. Targeting histone methyltransferase EZH2 as cancer treatment. J. Biochem. 2014, 156, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Garapaty-Rao, S.; Nasveschuk, C.; Gagnon, A.; Chan, E.Y.; Sandy, P.; Busby, J.; Balasubramanian, S.; Campbell, R.; Zhao, F.; Bergeron, L.; et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem. Biol. 2013, 20, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, V.; Iyer, P.; Arora, S.; Troyer, P.; Normant, E. Abstract 1697: CPI-169, a novel and potent EZH2 inhibitor, synergizes with CHOP in vivo and achieves complete regression in lymphoma xenograft models. Cancer Res. 2014, 74, 1697–1697. [Google Scholar] [CrossRef]
- Kung, P.-P.; Huang, B.; Zehnder, L.; Tatlock, J.; Bingham, P.; Krivacic, C.; Gajiwala, K.; Diehl, W.; Yu, X.; Maegley, K.A. SAH derived potent and selective EZH2 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1532–1537. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Wei, Y.; Huang, X.; Fu, R.; Luo, X.; Zhu, X.; Lei, W.; Fang, J.; Li, H.; Wen, W. Inhibition of GSK 3β activity is associated with excessive EZH2 expression and enhanced tumour invasion in nasopharyngeal carcinoma. PLoS ONE 2013, 8, e68614. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Ueki, N.; Cheng, J.; Imanishi, H.; Hada, T. Induction of apoptosis in hepatocellular carcinoma cell lines by emodin. Jpn. J. Cancer Res. 2002, 93, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.-L.; Chuang, M.-J.; Tang, S.-H.; Wu, S.-T.; Sun, K.-H.; Chen, T.-T.; Sun, G.-H.; Chang, S.-Y.; Yu, C.-P.; Ho, J.-Y.; et al. Emodin modulates epigenetic modifications and suppresses bladder carcinoma cell growth. Mol. Carcinog. 2015, 54, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Yu, L.; Li, Z.; Shen, Y.; Wang, J.; Cai, J.; Xiao, L.; Wang, Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol. Ther. 2010, 10, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Cai, J.; Ding, H.; Xu, L.; Yang, Q.; Wang, Z. EZH2 participates in malignant biological behavior of epithelial ovarian cancer through regulating the expression of BRCA1. Cancer Biol. Ther. 2014, 15, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Adelaiye-Ogala, R.M.; Chintala, S.; Shen, L.; Orillion, A.; Ciamporcero, E.; Elbanna, M.; Miles, K.M.; Gillard, B.; Buck, M.; Pili, R. Abstract 3508: Inhibition of EZH2 overcomes resistance to sunitinib in clear cell renal cell carcinoma models. Cancer Res. 2015, 75, 3508. [Google Scholar] [CrossRef]
- Lv, Y.; Yuan, C.; Xiao, X.; Wang, X.; Ji, X.; Yu, H.; Wu, Z.; Zhang, J. The expression and significance of the enhancer of zeste homolog 2 in lung adenocarcinoma. Oncol. Rep. 2012, 28, 147–154. [Google Scholar] [PubMed]
- Reijm, E.A.; Timmermans, A.M.; Look, M.P.; Meijer-van Gelder, M.E.; Stobbe, C.K.; van Deurzen, C.H.M.; Martens, J.W.M.; Sleijfer, S.; Foekens, J.A.; Berns, P.M.J.J.; et al. High protein expression of EZH2 is related to unfavorable outcome to tamoxifen in metastatic breast cancer. Ann. Oncol. 2014, 25, 2185–2190. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.-Y.; Wang, H.; Xiang, P.; Liu, Y.-W.; Li, H.-Z.; Lei, B.-X.; Yu, M.; Qi, S.-T. Inhibition of EZH2 reverses chemotherapeutic drug TMZ chemosensitivity in glioblastoma. Int. J. Clin. Exp. Pathol. 2014, 7, 6662–6670. [Google Scholar] [PubMed]
- De Vries, N.A.; Hulsman, D.; Akhtar, W.; de Jong, J.; Miles, D.C.; Blom, M.; van Tellingen, O.; Jonkers, J.; van Lohuizen, M. Prolonged EZH2 depletion in glioblastoma causes a robust switch in cell fate resulting in tumor progression. Cell. Rep. 2015, 10, 383–397. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Fernández, M.; Rubio, C.; Segovia, C.; López-Calderón, F.F.; Dueñas, M.; Paramio, J.M. EZH2 in Bladder Cancer, a Promising Therapeutic Target. Int. J. Mol. Sci. 2015, 16, 27107-27132. https://doi.org/10.3390/ijms161126000
Martínez-Fernández M, Rubio C, Segovia C, López-Calderón FF, Dueñas M, Paramio JM. EZH2 in Bladder Cancer, a Promising Therapeutic Target. International Journal of Molecular Sciences. 2015; 16(11):27107-27132. https://doi.org/10.3390/ijms161126000
Chicago/Turabian StyleMartínez-Fernández, Mónica, Carolina Rubio, Cristina Segovia, Fernando F. López-Calderón, Marta Dueñas, and Jesús M. Paramio. 2015. "EZH2 in Bladder Cancer, a Promising Therapeutic Target" International Journal of Molecular Sciences 16, no. 11: 27107-27132. https://doi.org/10.3390/ijms161126000
APA StyleMartínez-Fernández, M., Rubio, C., Segovia, C., López-Calderón, F. F., Dueñas, M., & Paramio, J. M. (2015). EZH2 in Bladder Cancer, a Promising Therapeutic Target. International Journal of Molecular Sciences, 16(11), 27107-27132. https://doi.org/10.3390/ijms161126000