
Sildenafil Protects against Myocardial Ischemia-Reperfusion Injury Following Cardiac Arrest in a Porcine Model: Possible Role of the Renin-Angiotensin System

Abstract
:
1. Introduction
2. Results
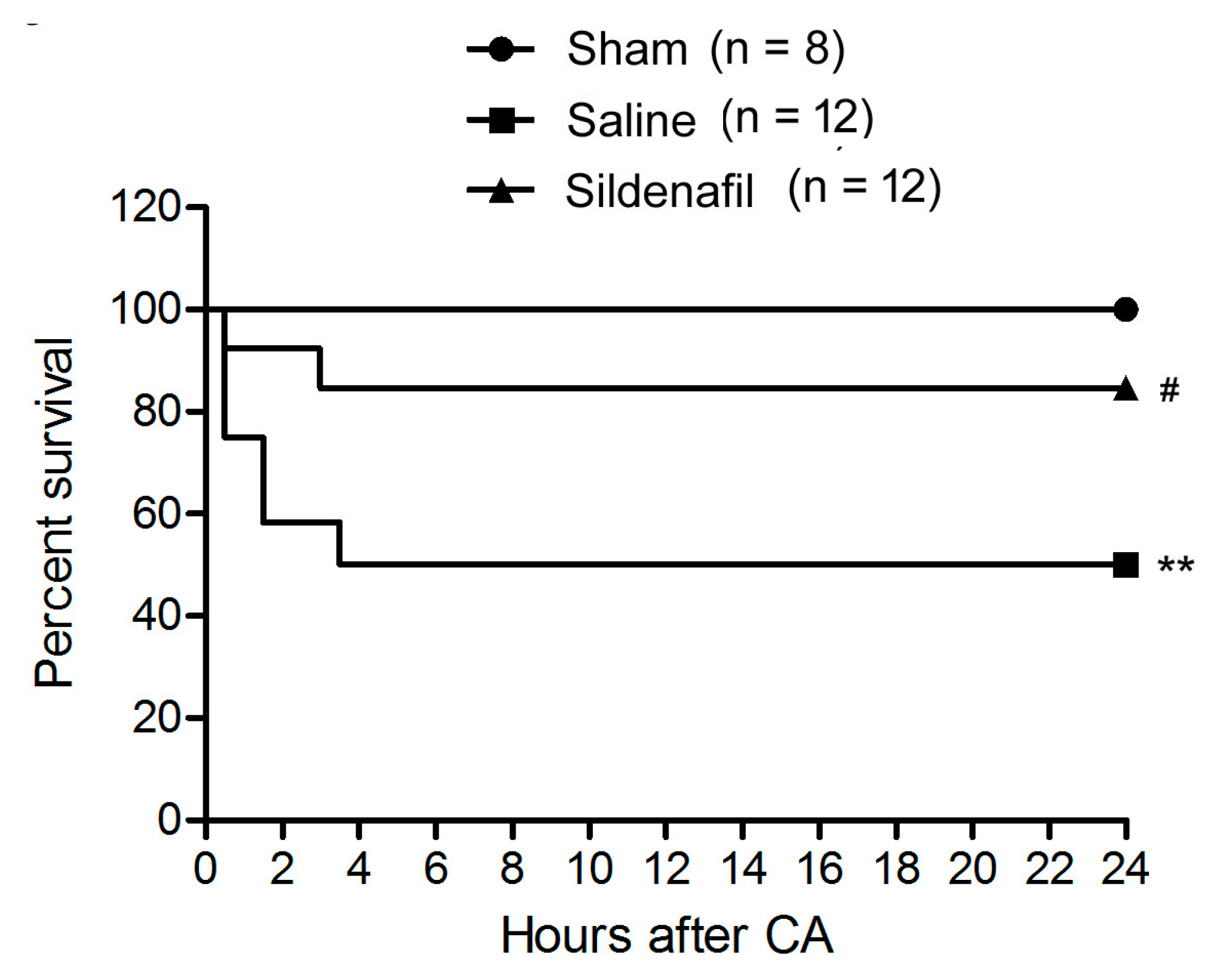
2.1. Sildenafil Improves Survival Rate in Pig CAR Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Outcomes | Sham (n = 8) | Saline (n = 12) | Sildenafil (n = 12) |
|---|---|---|---|
| Number of shocks | 0 | 4.9 ± 1.2 ** | 2.1 ± 0.6 #,** |
| Total adrenaline dose (mg) | 0 | 1.9 ± 0.7 ** | 0.8 ± 0.3 #,** |
| Energy of shock (J) | 0 | 260.5 ± 27.8 ** | 212.7 ± 24.2 #,** |
| Time to ROSC (min) | 0 | 6 ± 2.2 ** | 4 ± 1.3 ** |
| Number of surviving pigs (at ROSC) | 8 | 9 | 11 |
| Number of surviving pigs (6-h after ROSC) | 8 | 6 ** | 10 # |

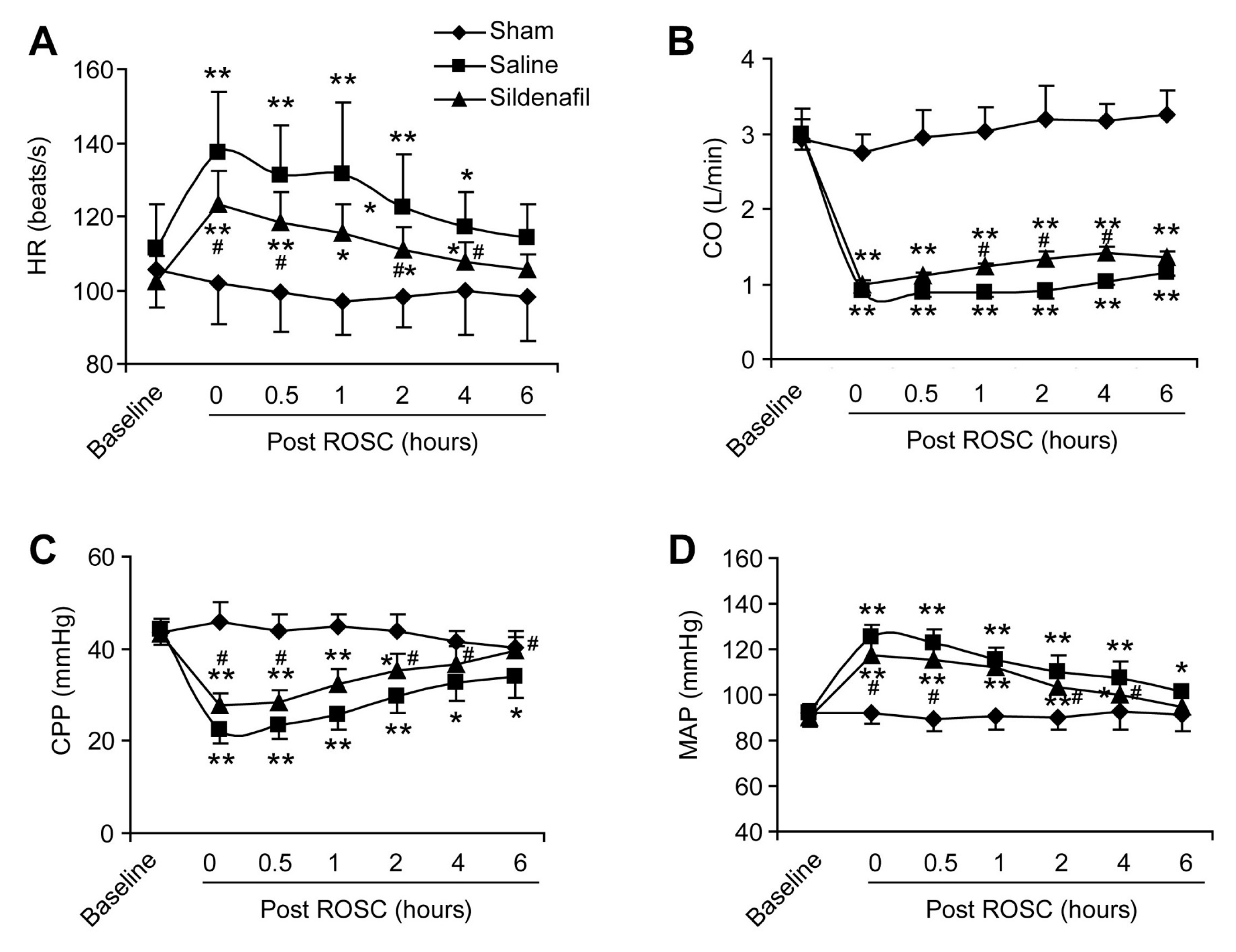
2.2. Sildenafil Ameliorates the Reduced Cardiac Function in Pig CAR Model

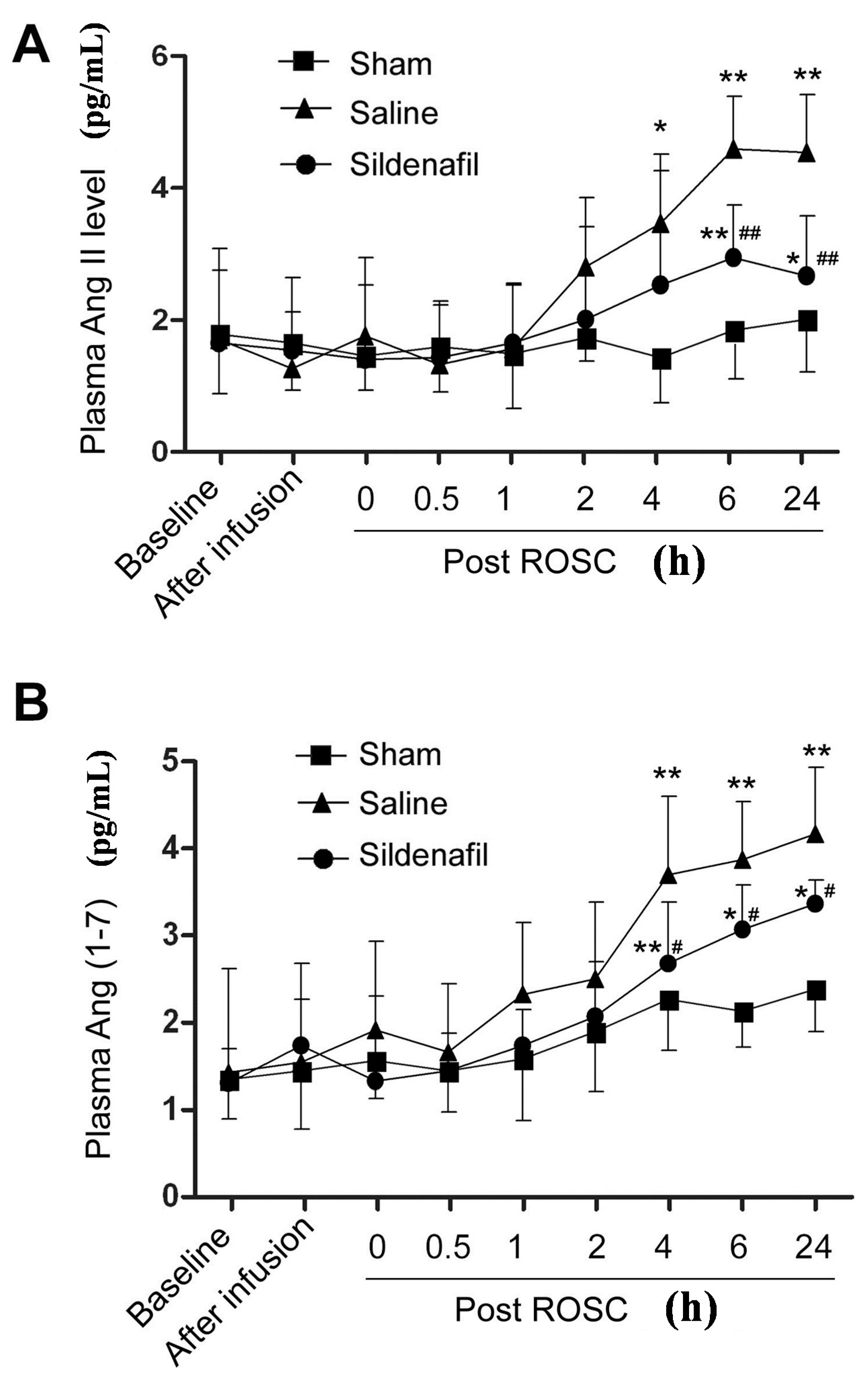
2.3. Plasma Ang II and Ang (1–7) Levels Are Decreased by Sildenafil in Pig CAR Model

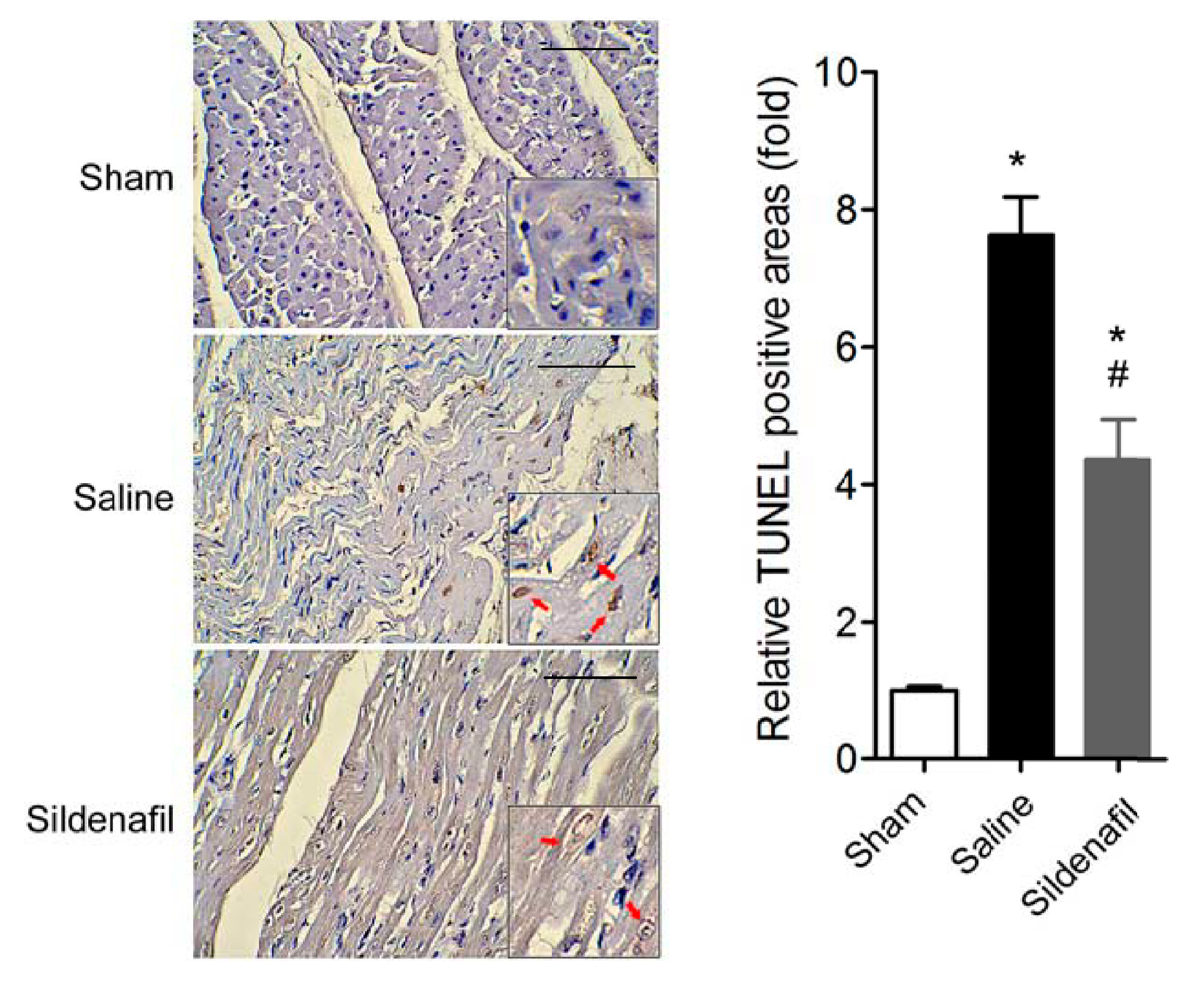
2.4. Sildenafil Attenuates Myocardial Apoptosis in Pig CAR Model

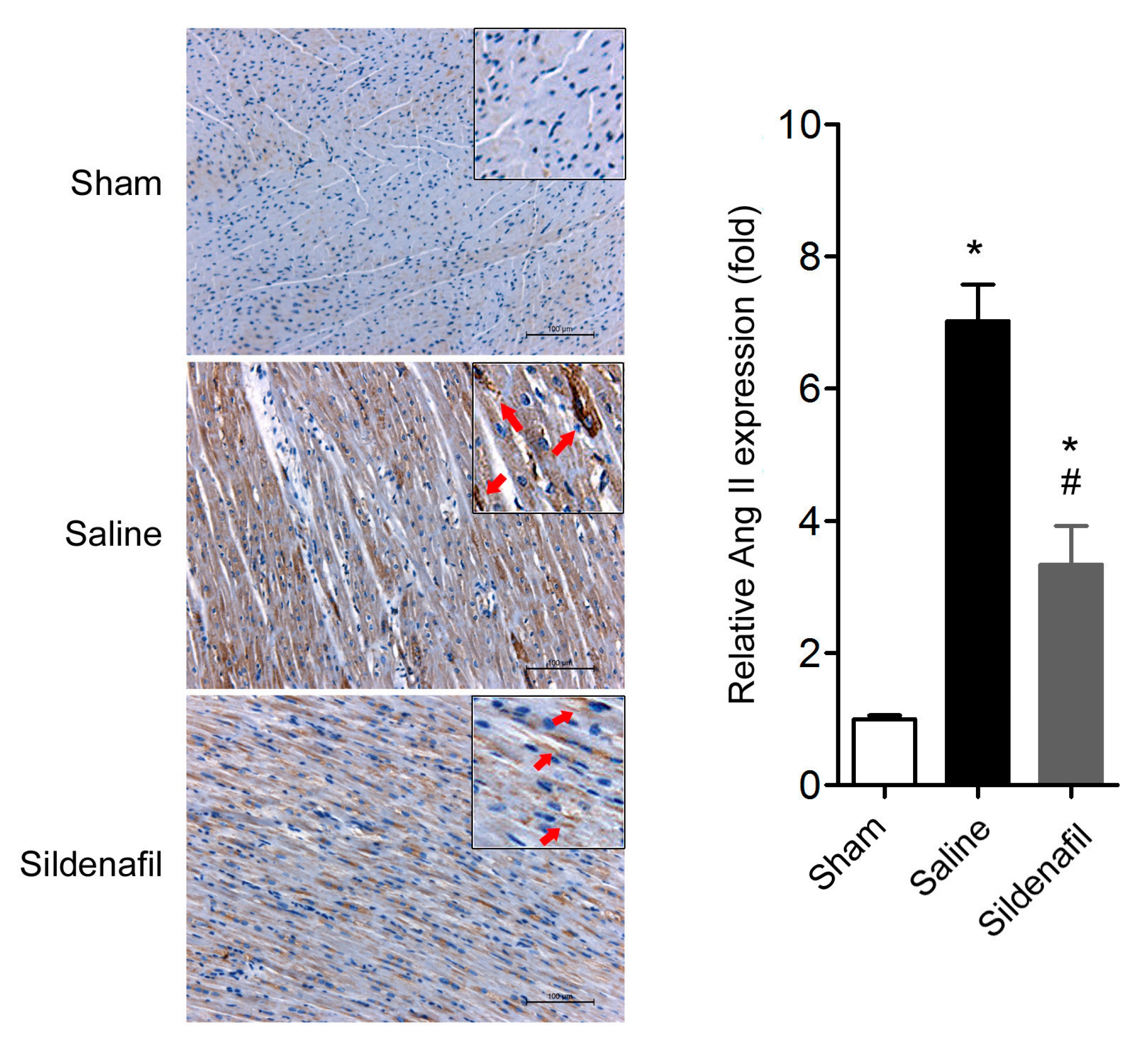
2.5. Sildenafil Reduces Myocardial Ang II Expression in Pig CAR Model

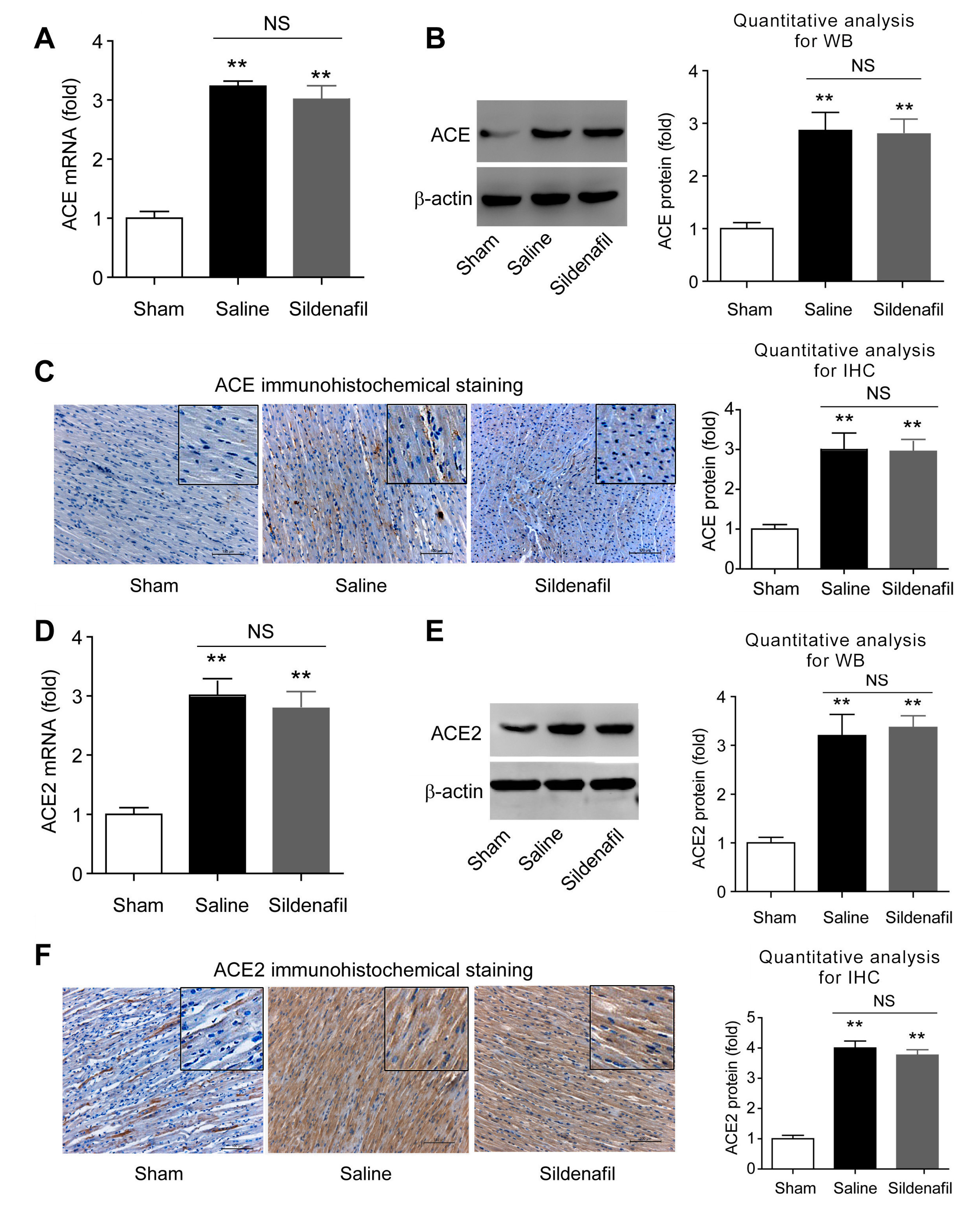
2.6. Sildenafil Does not Change ACE and ACE2 Expressions in Heart Tissue in Pig CAR Model

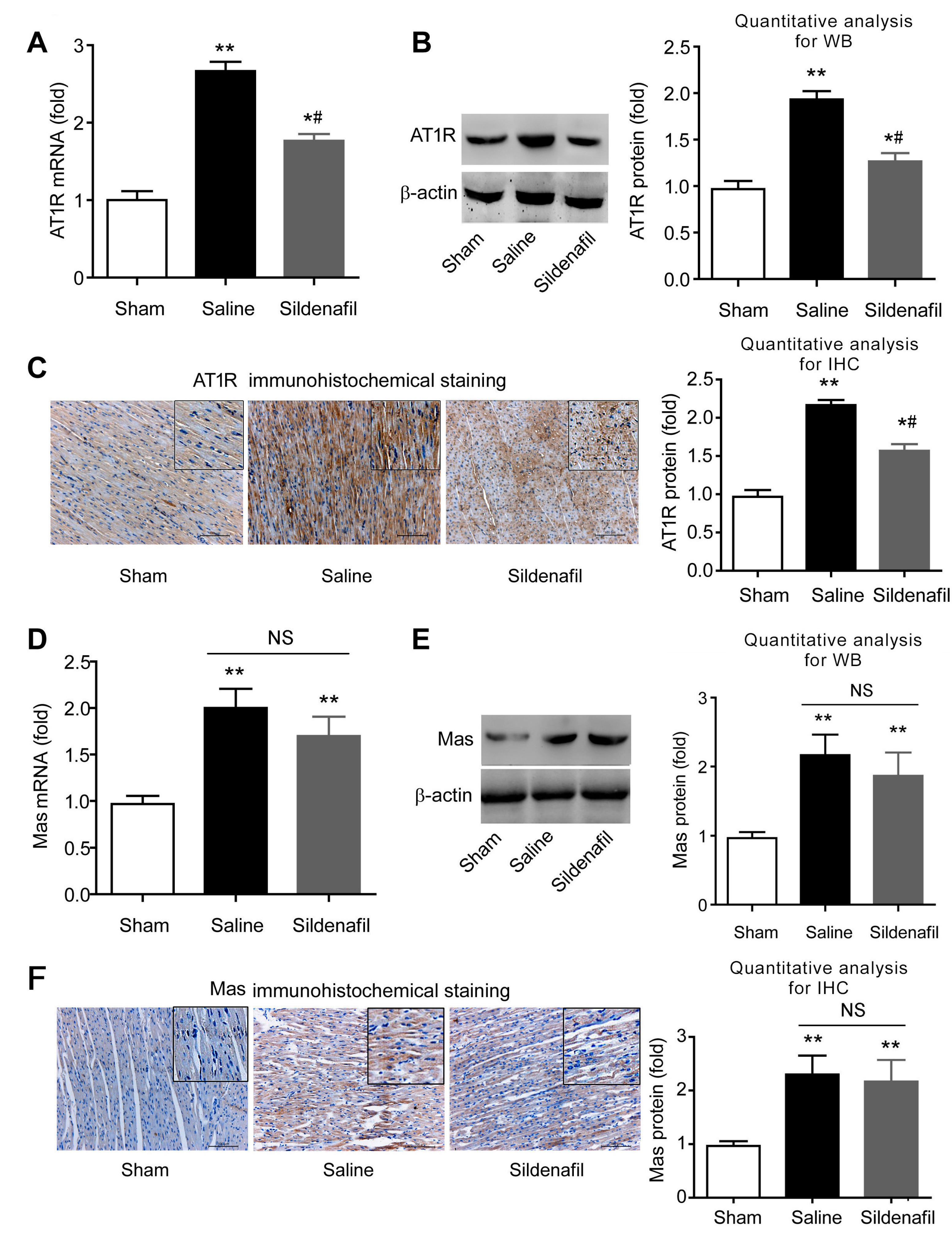
2.7. Sildenafil Decreases AT1R But Has no Effect on Mas Expression in Pig CAR Model

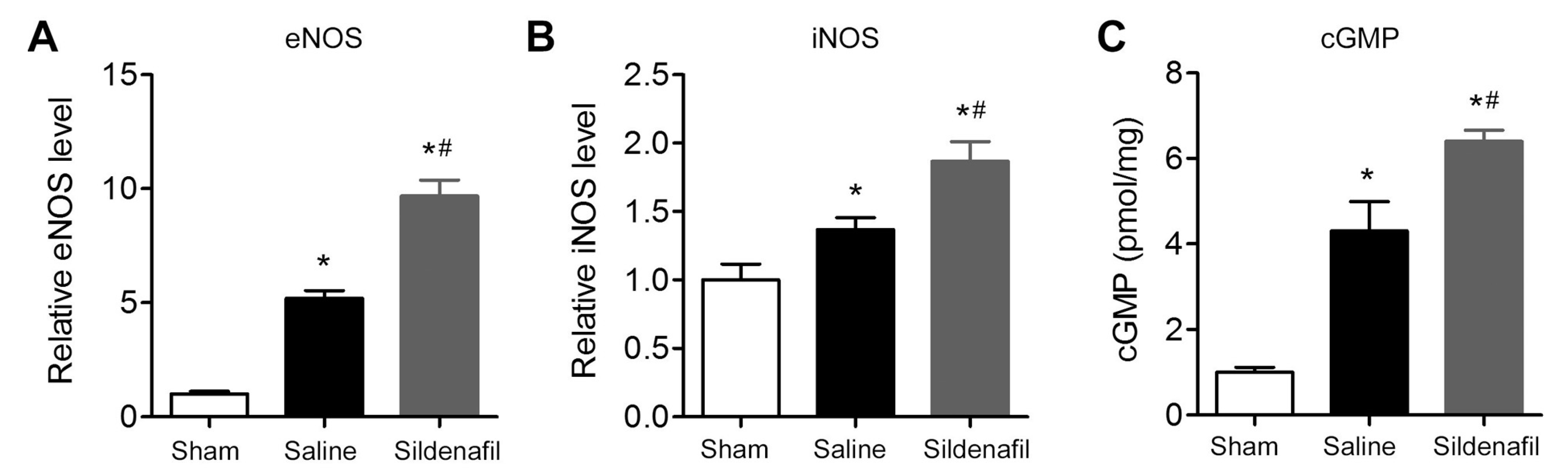
2.8. Sildenafil Increases eNOS, iNOS and cGMP Levels in Myocardial Tissue in Pig CAR Model

3. Discussion
4. Materials and Methods
4.1. Animals
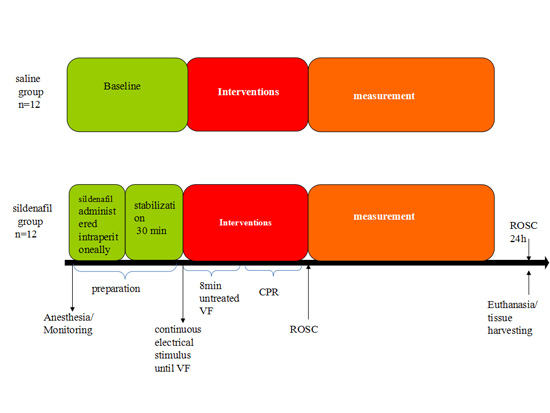
4.2. Experimental Procedures
4.3. Experimental Myocardial I/R Injury by CA
4.4. Hemodynamic Parameters
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Quantitative Real-Time PCR (qRT-PCR)
4.7. Immunohistochemical Staining
4.8. Western Blot Analysis
4.9. TUNEL Assay
4.10. Measurement of cGMP Levels
4.11. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Perkins, G.D.; Jacobs, I.G.; Nadkarni, V.M.; Berg, R.A.; Bhanji, F.; Biarent, D.; Bossaert, L.L.; Brett, S.J.; Chamberlain, D.; de Caen, A.R.; et al. Cardiac arrest and cardiopulmonary resuscitation outcome reports: Update of the utstein resuscitation registry templates for out-of-hospital cardiac arrest: A statement for healthcare professionals from a task force of the international liaison committee on resuscitation (american heart association, european resuscitation council, australian and new zealand council on resuscitation, heart and stroke foundation of canada, interamerican heart foundation, resuscitation council of southern africa, resuscitation council of asia); and the american heart association emergency cardiovascular care committee and the council on cardiopulmonary, critical care, perioperative and resuscitation. Circulation 2015, 132, 1286–1300. [Google Scholar] [PubMed]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Diep, Q.N.; Benkirane, K.; Amiri, F.; Cohn, J.S.; Endemann, D.; Schiffrin, E.L. PPARα activator fenofibrate inhibits myocardial inflammation and fibrosis in angiotensin ii-infused rats. J. Mol. Cell. Cardiol. 2004, 36, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Sovari, A.A.; Iravanian, S.; Dolmatova, E.; Jiao, Z.; Liu, H.; Zandieh, S.; Kumar, V.; Wang, K.; Bernstein, K.E.; Bonini, M.G.; et al. Inhibition of c-SRC tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J. Am. Coll. Cardiol. 2011, 58, 2332–2339. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.J.; Santos, R.A.; Almeida, A.P. Angiotensin-(1–7): Cardioprotective effect in myocardial ischemia/reperfusion. Hypertension 2001, 38, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Lue, T.F.; Padma-Nathan, H.; Rosen, R.C.; Steers, W.D.; Wicker, P.A. Oral sildenafil in the treatment of erectile dysfunction. N. Engl. J. Med. 1998, 338, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; Vicenzi, M.; Arena, R.; Guazzi, M.D. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: Results of a 1-year, prospective, randomized, placebo-controlled study. Circ. Heart Fail. 2011, 4, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yuan, W.; Wang, G.; Wu, J.; Wang, M.; Li, C. The protective effects of phosphodiesterase-5 inhibitor, sildenafil on post-resuscitation cardiac dysfunction of cardiac arrest: Metabolic evidence from microdialysis. Crit. Care 2014, 18, 641. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Maulik, N.; Das, D.K.; Kadowitz, P.J.; Bivalacqua, T.J. Cardioprotection with sildenafil, a selective inhibitor of cyclic 3ʹ,5ʹ-monophosphate-specific phosphodiesterase 5. Drugs Exp. Clin. Res. 2002, 28, 213–219. [Google Scholar] [PubMed]
- Fisher, P.W.; Salloum, F.; Das, A.; Hyder, H.; Kukreja, R.C. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation 2005, 111, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Giannetta, E.; Isidori, A.M.; Galea, N.; Carbone, I.; Mandosi, E.; Vizza, C.D.; Naro, F.; Morano, S.; Fedele, F.; Lenzi, A. Chronic inhibition of cGMP phosphodiesterase 5A improves diabetic cardiomyopathy: A randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging. Circulation 2012, 125, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Giannetta, E.; Feola, T.; Gianfrilli, D.; Pofi, R.; Dall’Armi, V.; Badagliacca, R.; Barbagallo, F.; Lenzi, A.; Isidori, A.M. Is chronic inhibition of phosphodiesterase type 5 cardioprotective and safe? A meta-analysis of randomized controlled trials. BMC Med. 2014, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.M.; Cornacchione, M.; Barbagallo, F.; di Grazia, A.; Barrios, F.; Fassina, L.; Monaco, L.; Giannetta, E.; Gianfrilli, D.; Garofalo, S.; et al. Inhibition of type 5 phosphodiesterase counteracts β2-adrenergic signalling in beating cardiomyocytes. Cardiovasc. Res. 2015, 106, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Xi, L.; Kukreja, R.C. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J. Biol. Chem. 2005, 280, 12944–12955. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Salloum, F.N.; Xi, L.; Rao, Y.J.; Kukreja, R.C. Erk phosphorylation mediates sildenafil-induced myocardial protection against ischemia-reperfusion injury in mice. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1236–H1243. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Ockaili, R.; Salloum, F.; Kukreja, R.C. Protein kinase c plays an essential role in sildenafil-induced cardioprotection in rabbits. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1455–H1460. [Google Scholar] [CrossRef] [PubMed]
- Fibbi, B.; Morelli, A.; Marini, M.; Zhang, X.H.; Mancina, R.; Vignozzi, L.; Filippi, S.; Chavalmane, A.; Silvestrini, E.; Colli, E.; et al. Atorvastatin but not elocalcitol increases sildenafil responsiveness in spontaneously hypertensive rats by regulating the RhoA/ROCK pathway. J. Androl. 2008, 29, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.J.; Ersboll, M.; Axelsson, A.; Gustafsson, F.; Hassager, C.; Kober, L.; Borlaug, B.A.; Boesgaard, S.; Skovgaard, L.T.; Moller, J.E. Sildenafil and diastolic dysfunction after acute myocardial infarction in patients with preserved ejection fraction: SIDAMI. Circulation 2013, 127, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Salloum, F.N.; Abbate, A.; Das, A.; Houser, J.E.; Mudrick, C.A.; Qureshi, I.Z.; Hoke, N.N.; Roy, S.K.; Brown, W.R.; Prabhakar, S.; et al. Sildenafil (Viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1398–H1406. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Li, E.; Mu, Q.; Zhang, Y.; Wei, X.; Li, H.; Cheng, L.; Zhang, B. Hydrogen sulfide inhalation decreases early blood-brain barrier permeability and brain edema induced by cardiac arrest and resuscitation. J. Cereb. Blood Flow Metab. 2015, 35, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Zarychanski, R.; Abou-Setta, A.M.; Turgeon, A.F.; Houston, B.L.; McIntyre, L.; Marshall, J.C.; Fergusson, D.A. Association of hydroxyethyl starch administration with mortality and acute kidney injury in critically ill patients requiring volume resuscitation: A systematic review and meta-analysis. JAMA 2013, 309, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Zreikat, H.H.; Harpe, S.E.; Slattum, P.W.; Mays, D.P.; Essah, P.A.; Cheang, K.I. Effect of renin-angiotensin system inhibition on cardiovascular events in older hypertensive patients with metabolic syndrome. Metab. Clin. Exp. 2014, 63, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Capasso, J.M.; Huang, H.E.; Meggs, L.G.; Li, P.; Anversa, P. Ang II receptors, c-myc, and c-jun in myocytes after myocardial infarction and ventricular failure. Am. J. Physiol. 1993, 264, H760–H769. [Google Scholar] [PubMed]
- Wu, B.; Lin, R.; Dai, R.; Chen, C.; Wu, H.; Hong, M. Valsartan attenuates oxidative stress and nf-kappab activation and reduces myocardial apoptosis after ischemia and reperfusion. Eur. J. Pharmacol. 2013, 705, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Van Kats, J.P.; Duncker, D.J.; Haitsma, D.B.; Schuijt, M.P.; Niebuur, R.; Stubenitsky, R.; Boomsma, F.; Schalekamp, M.A.; Verdouw, P.D.; Danser, A.H. Angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade prevent cardiac remodeling in pigs after myocardial infarction: Role of tissue angiotensin II. Circulation 2000, 102, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Sugaya, T.; Murakami, K.; Yazaki, Y.; Komuro, I. Angiotensin II type 1A receptor knockout mice display less left ventricular remodeling and improved survival after myocardial infarction. Circulation 1999, 100, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.; Davie, A.P. Angiotensin-(1-7) attenuates the development of heart failure after myocardial infarction in rats. Circulation 2002, 106, e147. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.J.; Jacoby, B.A.; Araujo, C.A.; Macedo, F.A.; Silva, G.A.; Almeida, A.P.; Caliari, M.V.; Santos, R.A. The nonpeptide angiotensin-(1-7) receptor Mas agonist AVE-0991 attenuates heart failure induced by myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1113–H1119. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.T.; Cintra, A.S.; Frossard, J.C.; Palomino, Z.; Casarini, D.E.; Gomes, I.B.; Balarini, C.M.; Gava, A.L.; Campagnaro, B.P.; Pereira, T.M.; et al. Inhibition of phosphodiesterase 5 restores endothelial function in renovascular hypertension. J. Transl. Med. 2014, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Straubinger, J.; Schottle, V.; Bork, N.; Subramanian, H.; Dunnes, S.; Russwurm, M.; Gawaz, M.; Friebe, A.; Nemer, M.; Nikolaev, V.O.; et al. Sildenafil does not prevent heart hypertrophy and fibrosis induced by cardiomyocyte angiotensin II type 1 receptor signaling. J. Pharmacol. Exp. Ther. 2015, 354, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Salloum, F.; Yin, C.; Xi, L.; Kukreja, R.C. Sildenafil induces delayed preconditioning through inducible nitric oxide synthase-dependent pathway in mouse heart. Circ. Res. 2003, 92, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, O.; Qin, Q.; Krieg, T.; Yang, X.M.; Philipp, S.; Critz, S.D.; Cohen, M.V.; Downey, J.M. Bradykinin induces mitochondrial ROS generation via NO, cGMP, pKG, and mitoKatp channel opening and leads to cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H468–H476. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Greer, J.J.; Lefer, D.J. Sildenafil-mediated acute cardioprotection is independent of the NO/cGMP pathway. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H342–H347. [Google Scholar] [CrossRef] [PubMed]
- Shalwala, M.; Zhu, S.G.; Das, A.; Salloum, F.N.; Xi, L.; Kukreja, R.C. Sirtuin 1 (Sirt1) activation mediates sildenafil induced delayed cardioprotection against ischemia-reperfusion injury in mice. PLoS ONE 2014, 9, e86977. [Google Scholar] [CrossRef] [PubMed]
- Madhani, M.; Hall, A.R.; Cuello, F.; Charles, R.L.; Burgoyne, J.R.; Fuller, W.; Hobbs, A.J.; Shattock, M.J.; Eaton, P. Phospholemman Ser69 phosphorylation contributes to sildenafil-induced cardioprotection against reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H827–H836. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.A.; Hlaing, S.M.; Gutierrez, R.A.; Sanchez, M.D.; Kovanecz, I.; Artaza, J.N.; Ferrini, M.G. Sildenafil attenuates inflammation and oxidative stress in pelvic ganglia neurons after bilateral cavernosal nerve damage. Int. J. Mol. Sci. 2014, 15, 17204–17220. [Google Scholar] [CrossRef] [PubMed]
- Venneri, M.A.; Giannetta, E.; Panio, G.; de Gaetano, R.; Gianfrilli, D.; Pofi, R.; Masciarelli, S.; Fazi, F.; Pellegrini, M.; Lenzi, A.; et al. Chronic inhibition of pde5 limits pro-inflammatory monocyte-macrophage polarization in streptozotocin-induced diabetic mice. PLoS ONE 2015, 10, e0126580. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Gonon, A.T.; Sjöquist, P.-O.; Lundberg, J.O.; Pernow, J. Arginase inhibition mediates cardioprotection during ischaemia-reperfusion. Cardiovasc. Res. 2010, 85, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, T.; Akiyama, T.; Du, C.K.; Zhan, D.Y.; Shirai, M. Contribution of calpain to myoglobin efflux from cardiomyocytes during ischaemia and after reperfusion in anaesthetized rats. Acta Physiol. 2014, 210, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Liepinsh, E.; Makrecka, M.; Kuka, J.; Makarova, E.; Vilskersts, R.; Cirule, H.; Sevostjanovs, E.; Grinberga, S.; Pugovics, O.; Dambrova, M. The heart is better protected against myocardial infarction in the fed state compared to the fasted state. Metab. Clin. Exp. 2014, 63, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.K.; Jackson, E.K.; Luscher, T.F. Nitric oxide inhibits angiotensin ii-induced migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. J. Clin. Investig. 1995, 96, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yu, X.; Brecher, P. Nitric oxide inhibits angiotensin II-induced activation of the calcium-sensitive tyrosine kinase proline-rich tyrosine kinase 2 without affecting epidermal growth factor receptor transactivation. J. Biol. Chem. 1999, 274, 24342–24348. [Google Scholar] [CrossRef] [PubMed]
- Cahill, P.A.; Redmond, E.M.; Foster, C.; Sitzmann, J.V. Nitric oxide regulates angiotensin II receptors in vascular smooth muscle cells. Eur. J. Pharmacol. 1995, 288, 219–229. [Google Scholar] [CrossRef]
- Ichiki, T.; Usui, M.; Kato, M.; Funakoshi, Y.; Ito, K.; Egashira, K.; Takeshita, A. Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension 1998, 31, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Ichiki, T.; Katoh, M.; Egashira, K.; Takeshita, A. Regulation of angiotensin II receptor expression by nitric oxide in rat adrenal gland. Hypertension 1998, 32, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.M.; Zheng, H.; Li, Y.F.; Patel, K.P. Nitric oxide inhibits the expression of at1 receptors in neurons. Am. J. Physiol. Cell Physiol. 2012, 302, C1162–C1173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, G.; Yuan, W.; Wu, J.; Wang, M.; Li, C. The effects of phosphodiesterase-5 inhibitor sildenafil against post-resuscitation myocardial and intestinal microcirculatory dysfunction by attenuating apoptosis and regulating micrornas expression: Essential role of nitric oxide syntheses signaling. J. Transl. Med. 2015, 13, 177. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.J.; Li, C.S.; Yin, W.P.; Hou, X.M.; Gu, W.; Zhang, D. Comparison of shock-first strategy and cardiopulmonary resuscitation-first strategy in a porcine model of prolonged cardiac arrest. Resuscitation 2013, 84, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, C.; Ji, X.; Yang, L.; Su, Z.; Wu, J. Effect of continuous compressions and 30:2 cardiopulmonary resuscitation on global ventilation/perfusion values during resuscitation in a porcine model. Crit. Care Med. 2010, 38, 2024–2030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, C. Combination of epinephrine with esmolol attenuates post-resuscitation myocardial dysfunction in a porcine model of cardiac arrest. PLoS ONE 2013, 8, e82677. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.F.; Li, C.S.; Wang, S.; Yang, L.; Cong, L.H. Comparison of the efficacy of nifekalant and amiodarone in a porcine model of cardiac arrest. Resuscitation 2010, 81, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, T.D.; Roe, D.J.; Cretin, S.; Spaite, D.W.; Larsen, M.P. Estimating effectiveness of cardiac arrest interventions: A logistic regression survival model. Circulation 1997, 96, 3308–3313. [Google Scholar] [CrossRef] [PubMed]
- Kern, K.B.; Hilwig, R.W.; Berg, R.A.; Sanders, A.B.; Ewy, G.A. Importance of continuous chest compressions during cardiopulmonary resuscitation: Improved outcome during a simulated single lay-rescuer scenario. Circulation 2002, 105, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, T.Y.; Wei, K.; Guan, Y.F.; Wang, X.; Xu, H.; Su, D.F.; Pei, G.; Miao, C.Y. Arrb1/β-arrestin-1 mediates neuroprotection through coordination of becn1-dependent autophagy in cerebral ischemia. Autophagy 2014, 10, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, T.Y.; Guan, Y.F.; Zhao, Y.; Li, Z.Y.; Lan, X.H.; Wang, X.; Yang, P.Y.; Kang, Z.M.; Vanhoutte, P.M.; et al. Vascular smooth muscle cell apoptosis is an early trigger for hypothyroid atherosclerosis. Cardiovasc. Res. 2014, 102, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, T.Y.; Guan, Y.F.; Tian, W.W.; Viollet, B.; Rui, Y.C.; Zhai, Q.W.; Su, D.F.; Miao, C.Y. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through sirt1-dependent adenosine monophosphate-activated kinase pathway. Ann. Neurol. 2011, 69, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhang, Q.; Yin, W.; Li, C. Caspase-3-mediated splenic lymphocyte apoptosis in a porcine model of cardiac arrest. Am. J. Emerg. Med. 2014, 32, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, T.Y.; Guan, Y.F.; Su, D.F.; Fan, G.R.; Miao, C.Y. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: Role of nicotinamide mononucleotide. Cardiovasc. Res. 2009, 81, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Du, H.; Zhou, C.-C.; Song, J.; Liu, X.; Cao, X.; Mehta, J.L.; Shi, Y.; Su, D.-F.; Miao, C.-Y. Intracellular NAMPT-NAD+-SIRT1 cascade improves post-ischaemic vascular repair by modulating notch signalling in endothelial progenitors. Cardiovasc. Res. 2014, 104, 477–488. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Zhang, Q.; Yuan, W.; Wu, J.; Li, C. Sildenafil Protects against Myocardial Ischemia-Reperfusion Injury Following Cardiac Arrest in a Porcine Model: Possible Role of the Renin-Angiotensin System. Int. J. Mol. Sci. 2015, 16, 27015-27031. https://doi.org/10.3390/ijms161126010
Wang G, Zhang Q, Yuan W, Wu J, Li C. Sildenafil Protects against Myocardial Ischemia-Reperfusion Injury Following Cardiac Arrest in a Porcine Model: Possible Role of the Renin-Angiotensin System. International Journal of Molecular Sciences. 2015; 16(11):27015-27031. https://doi.org/10.3390/ijms161126010
Chicago/Turabian StyleWang, Guoxing, Qian Zhang, Wei Yuan, Junyuan Wu, and Chunsheng Li. 2015. "Sildenafil Protects against Myocardial Ischemia-Reperfusion Injury Following Cardiac Arrest in a Porcine Model: Possible Role of the Renin-Angiotensin System" International Journal of Molecular Sciences 16, no. 11: 27015-27031. https://doi.org/10.3390/ijms161126010
APA StyleWang, G., Zhang, Q., Yuan, W., Wu, J., & Li, C. (2015). Sildenafil Protects against Myocardial Ischemia-Reperfusion Injury Following Cardiac Arrest in a Porcine Model: Possible Role of the Renin-Angiotensin System. International Journal of Molecular Sciences, 16(11), 27015-27031. https://doi.org/10.3390/ijms161126010