
Biodistribution, Stability, and Blood Distribution of the Cell Penetrating Peptide Maurocalcine in Mice

Abstract
:1. Introduction
2. Results
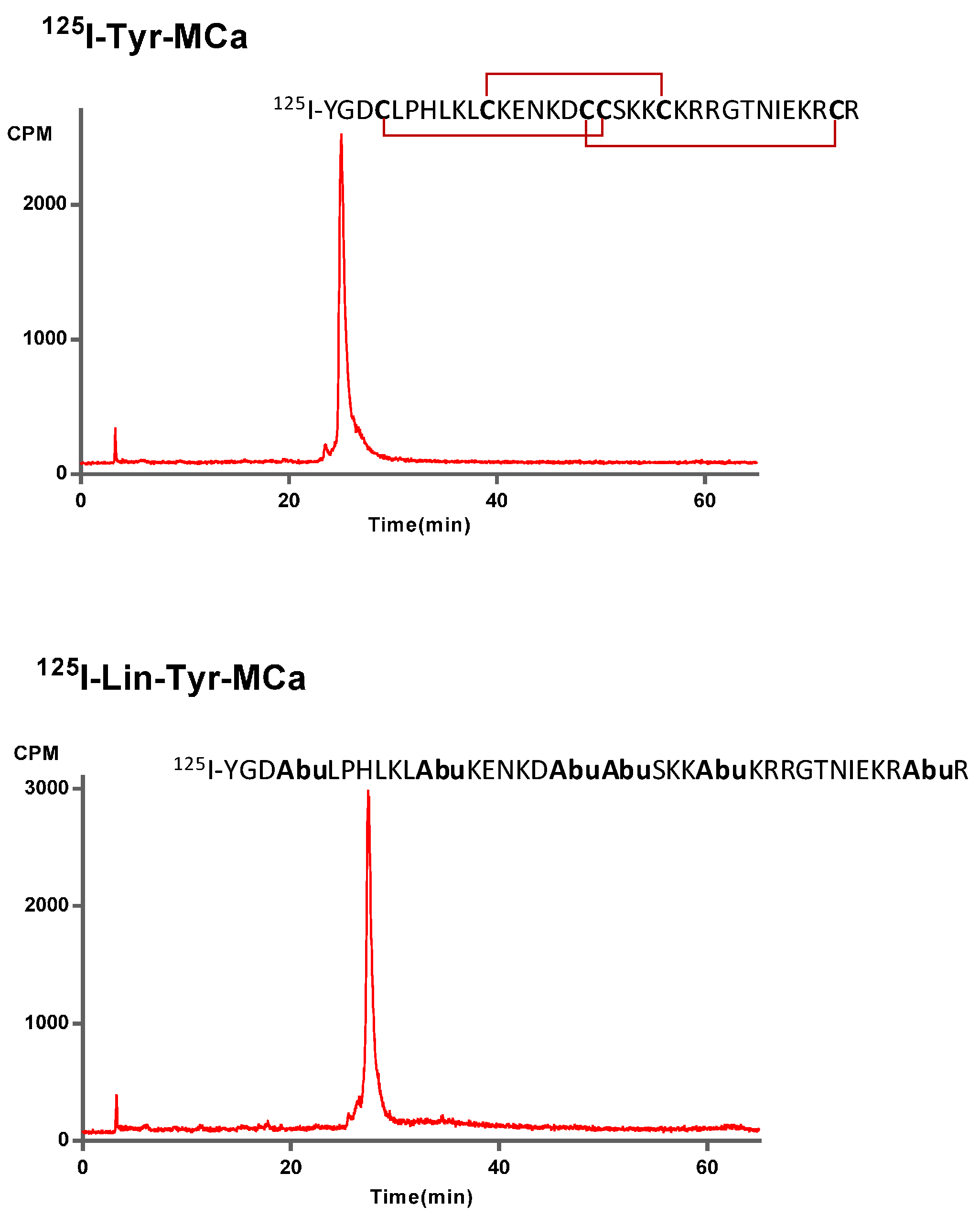
2.1. Chemical Synthesis and Radiolabeling
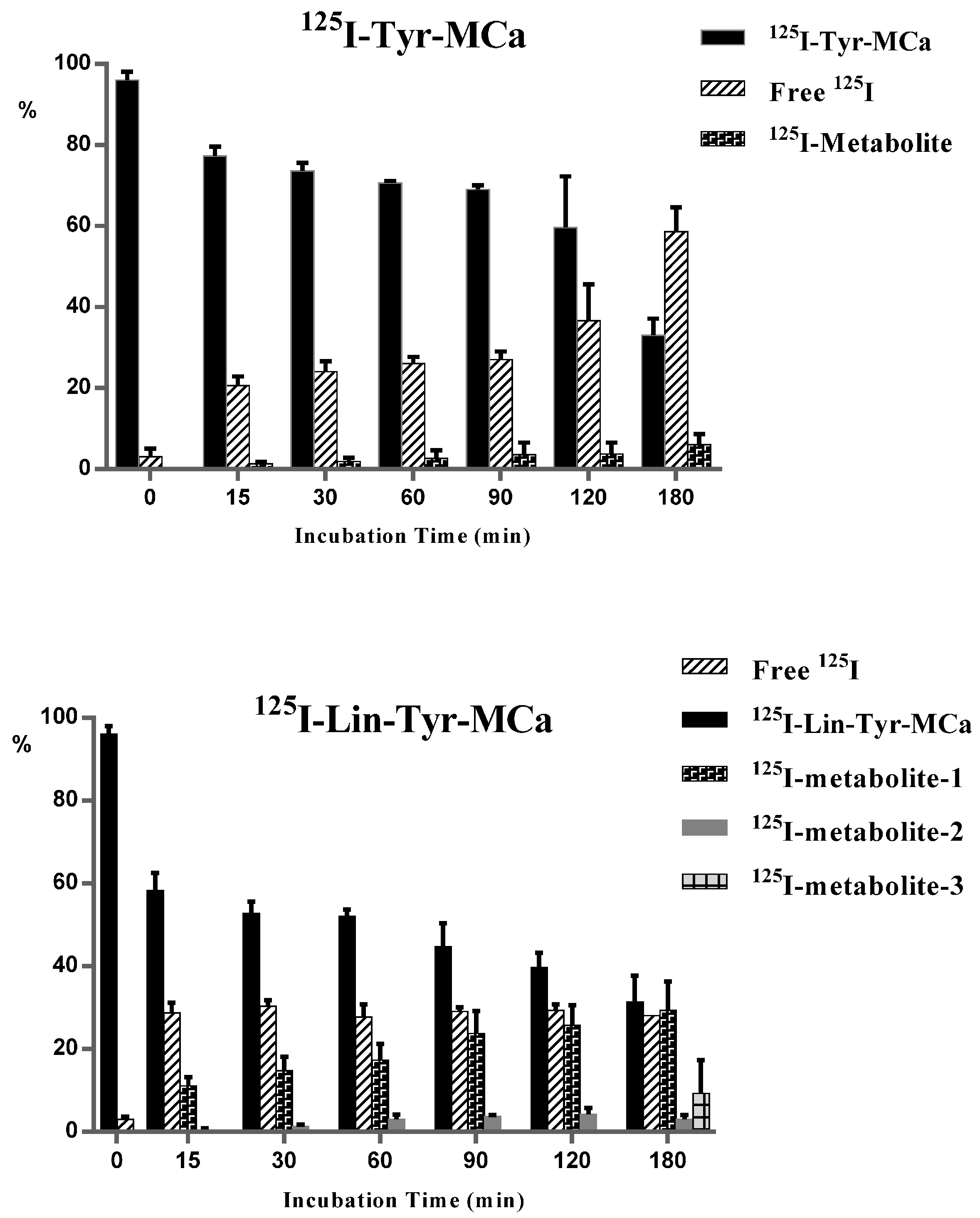
2.2. In Vitro Stability in Mouse Blood


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blood Distribution | 15 min | 30 min | 60 min | 90 min | 120 min | 180 min |
|---|---|---|---|---|---|---|
| 125I-Tyr-MCa | ||||||
| Blood cells | 22 ± 3 | 21 ± 3 | 22 ± 1 | 19 ± 3 | 18 ± 0 | 19 ± 2 |
| Plasma proteins | 64 ± 3 | 65 ± 5 | 64 ± 3 | 67 ± 3 | 69 ± 2 | 70 ± 2 |
| Protein-free plasma | 12 ± 4 | 12 ± 4 | 12 ± 3 | 12 ± 1 | 11 ± 1 | 9 ± 2 |
| 125I-Lin-Tyr-MCa | ||||||
| Blood cells | 30 ± 5 | 31 ± 8 | 35 ± 3 | 35 ± 9 | 37 ± 9 | 44 ± 10 |
| Plasma proteins | 47 ± 3 | 42 ± 3 | 34 ± 2 | 32 ± 0.4 | 34 ± 1 | 27 ± 5 |
| Protein-free plasma | 23 ± 4 | 23 ± 2 | 22 ± 3 | 30 ± 10 | 28 ± 8 | 28 ± 8 |

2.3. In Vivo Tracer Stability
| Blood Distribution | 15 min | 30 min | ||
|---|---|---|---|---|
| 125I-Tyr-MCa | Blood cells | 24 | 31 | |
| Plasma proteins | 47 | 42 | ||
| Protein-free plasma | 29 | 27 | ||
| Free 125I | 62 | 72 | ||
| 125I-Tyr-MCa | 38 | 28 |
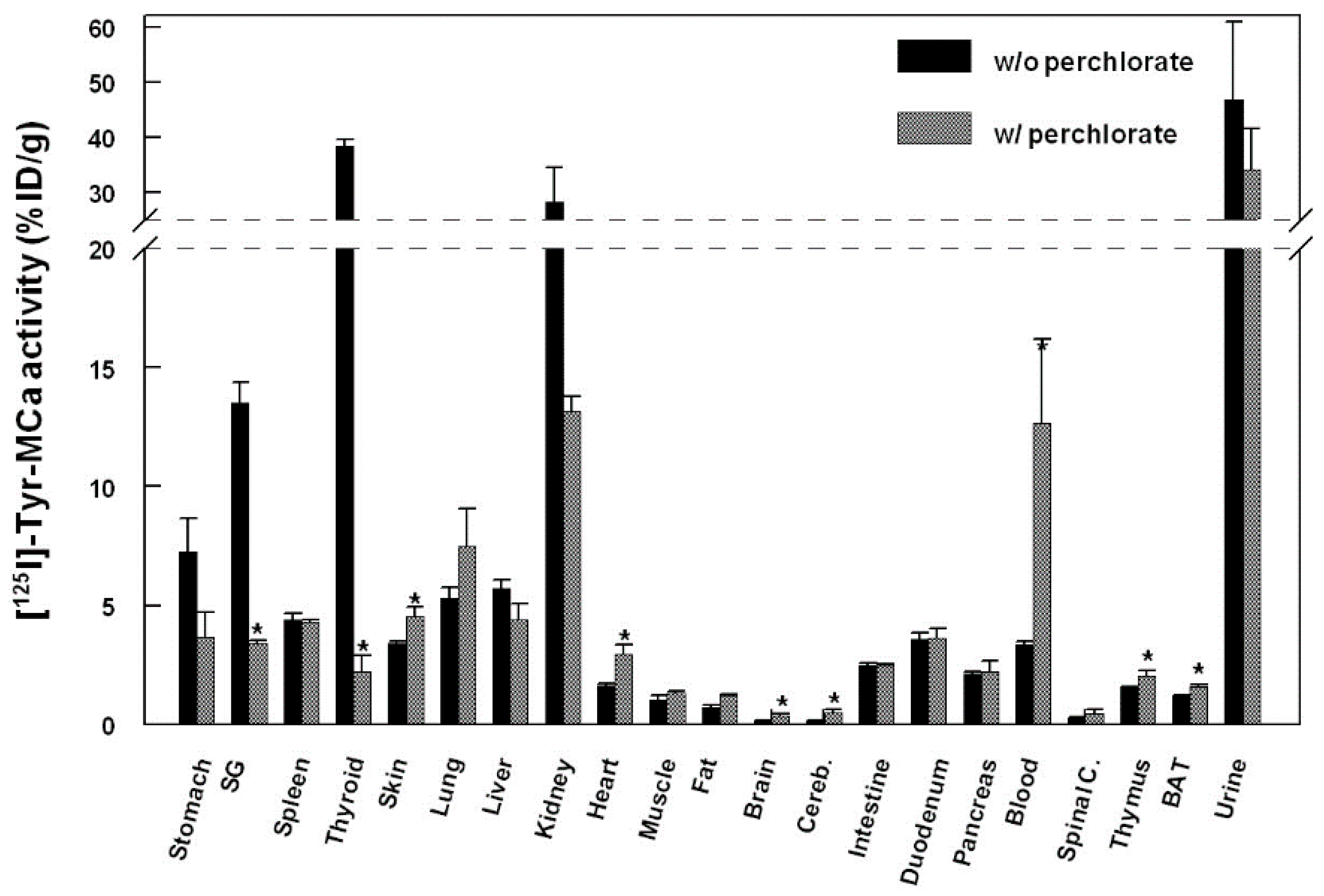
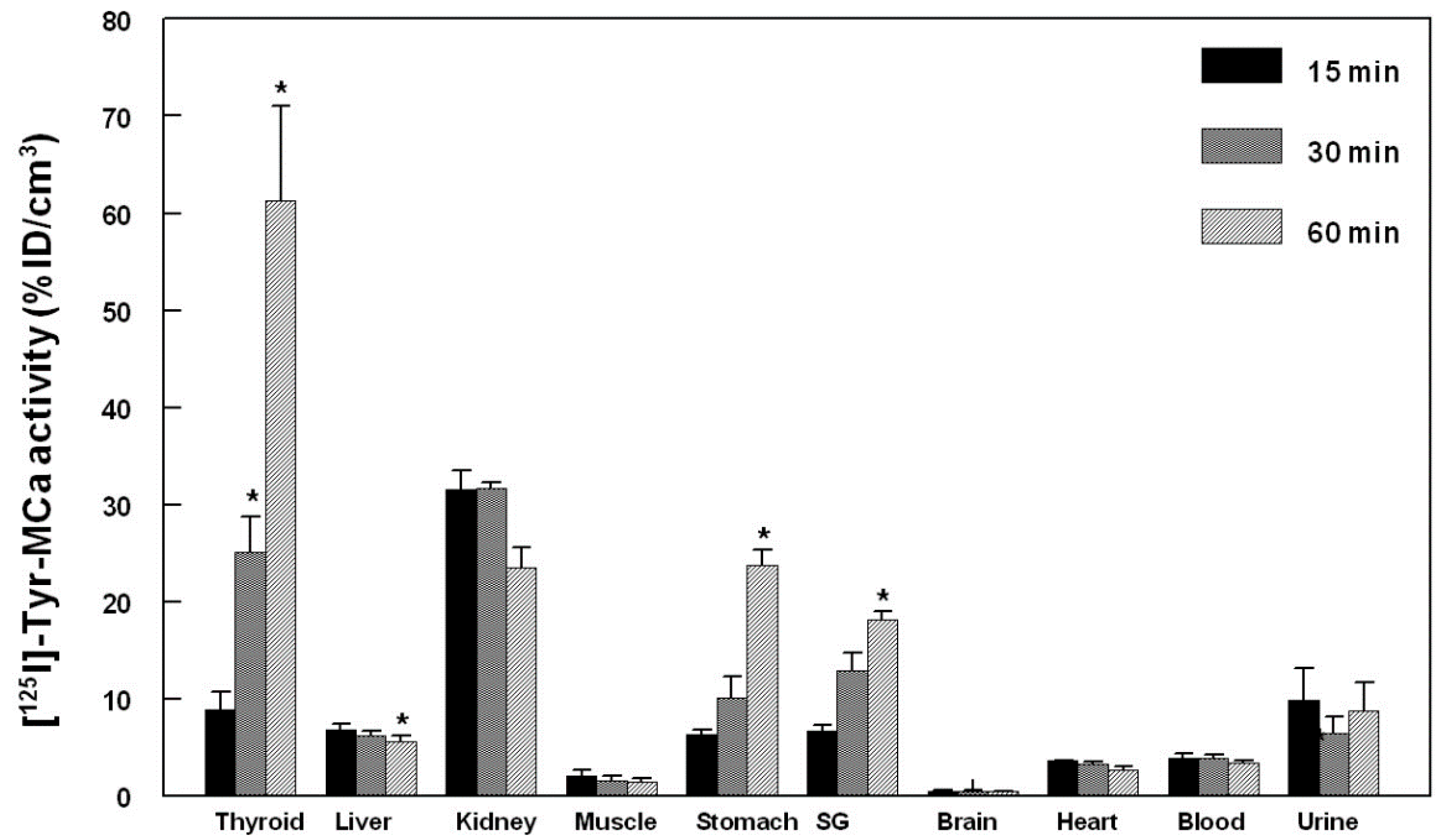
2.4. Biodistribution

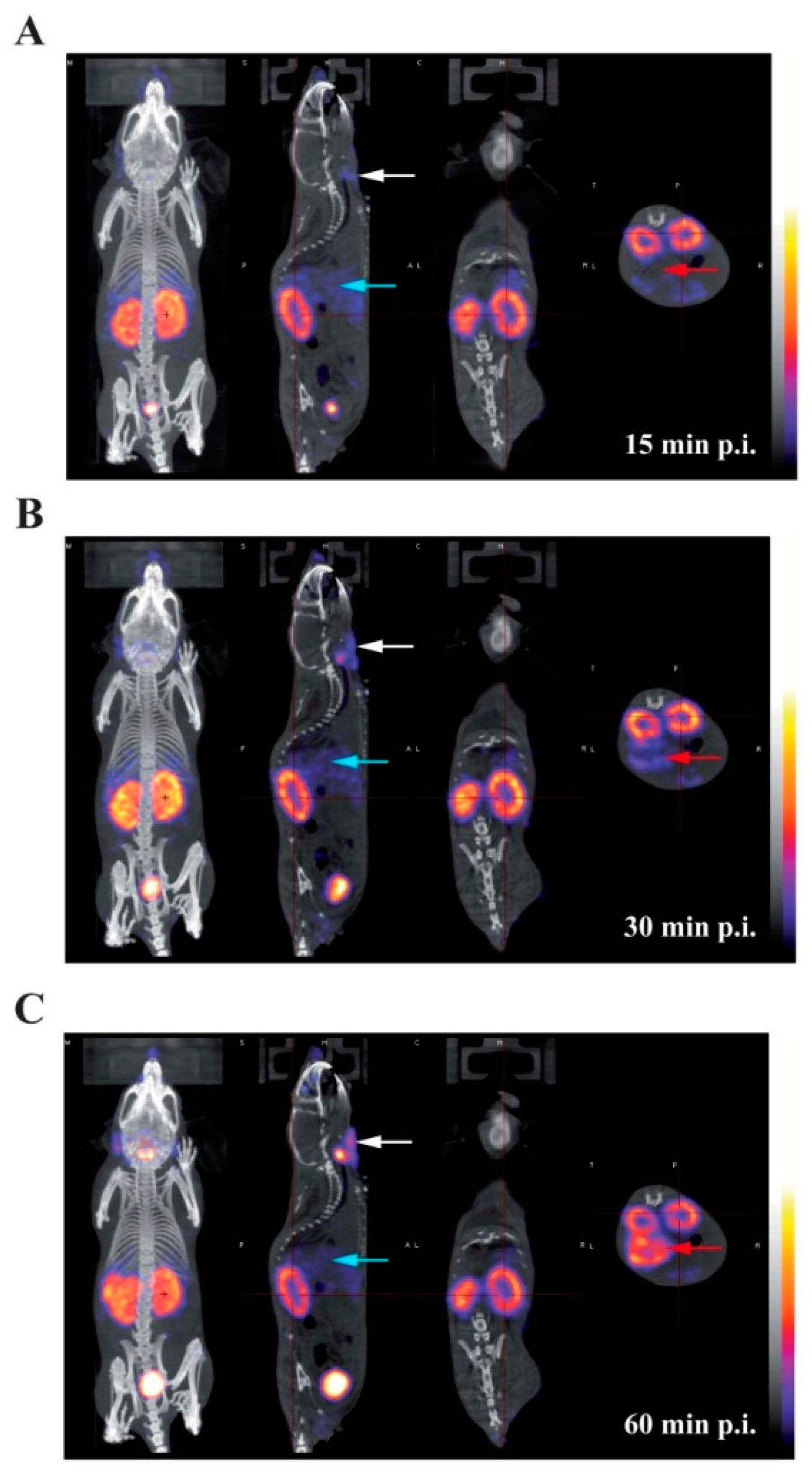
2.5. In Vivo Imaging


3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Chemical Synthesis and Radiolabeling
4.3. In Vitro Stability in Mouse Blood
4.4. In Vivo Experimental Protocol
4.4.1. Animals
4.4.2. In Vivo 125I-MCa Stability
4.4.3. Biodistribution
4.4.4. Post Mortem Analysis
4.5. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mosbah, A.; Kharrat, R.; Fajloun, Z.; Renisio, J.G.; Blanc, E.; Sabatier, J.M.; El Ayeb, M.; Darbon, H. A new fold in the scorpion toxin family, associated with an activity on a ryanodine-sensitive calcium channel. Proteins 2000, 40, 436–442. [Google Scholar] [CrossRef]
- Estève, E.; Mabrouk, K.; Dupuis, A.; Smida-Rezgui, S.; Altafaj, X.; Grunwald, D.; Platel, J.-C.; Andreotti, N.; Marty, I.; Sabatier, J.-M.; et al. Transduction of the scorpion toxin maurocalcine into cells. Evidence that the toxin crosses the plasma membrane. J. Biol. Chem. 2005, 280, 12833–12839. [Google Scholar] [CrossRef] [PubMed]
- Ram, N.; Aroui, S.; Jaumain, E.; Bichraoui, H.; Mabrouk, K.; Ronjat, M.; Lortat-Jacob, H.; de Waard, M. Direct peptide interaction with surface glycosaminoglycans contributes to the cell penetration of maurocalcine. J. Biol. Chem. 2008, 283, 24274–24284. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Ram, N.; Appaix, F.; Ronjat, M.; Kenani, A.; Pirollet, F.; de Waard, M. Maurocalcine as a non toxic drug carrier overcomes doxorubicin resistance in the cancer cell line MDA-MB 231. Pharm. Res. 2009, 26, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Boisseau, S.; Mabrouk, K.; Ram, N.; Garmy, N.; Collin, V.; Tadmouri, A.; Mikati, M.; Sabatier, J.-M.; Ronjat, M.; Fantini, J.; et al. Cell penetration properties of maurocalcine, a natural venom peptide active on the intracellular ryanodine receptor. Biochim. Biophys. Acta 2006, 1758, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Jayagopal, A.; Su, Y.R.; Blakemore, J.L.; Linton, M.F.; Fazio, S.; Haselton, F.R. Quantum dot mediated imaging of atherosclerosis. Nanotechnology 2009, 20, 165102. [Google Scholar] [CrossRef] [PubMed]
- Poillot, C.; Dridi, K.; Bichraoui, H.; Pêcher, J.; Alphonse, S.; Douzi, B.; Ronjat, M.; Darbon, H.; de Waard, M. D-Maurocalcine, a pharmacologically inert efficient cell-penetrating peptide analogue. J. Biol. Chem. 2010, 285, 34168–34180. [Google Scholar] [CrossRef] [PubMed]
- Ram, N.; Weiss, N.; Texier-Nogues, I.; Aroui, S.; Andreotti, N.; Pirollet, F.; Ronjat, M.; Sabatier, J.-M.; Darbon, H.; Jacquemond, V.; et al. Design of a disulfide-less, pharmacologically inert, and chemically competent analog of maurocalcine for the efficient transport of impermeant compounds into cells. J. Biol. Chem. 2008, 283, 27048–27056. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Brahim, S.; de Waard, M.; Bréard, J.; Kenani, A. Efficient induction of apoptosis by doxorubicin coupled to cell-penetrating peptides compared to unconjugated doxorubicin in the human breast cancer cell line MDA-MB 231. Cancer Lett. 2009, 285, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Brahim, S.; Hamelin, J.; de Waard, M.; Bréard, J.; Kenani, A. Conjugation of doxorubicin to cell penetrating peptides sensitizes human breast MDA-MB 231 cancer cells to endogenous TRAIL-induced apoptosis. Apoptosis Int. J. Program. Cell Death 2009, 14, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Brahim, S.; Waard, M.D.; Kenani, A. Cytotoxicity, intracellular distribution and uptake of doxorubicin and doxorubicin coupled to cell-penetrating peptides in different cell lines: A comparative study. Biochem. Biophys. Res. Commun. 2010, 391, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Mili, D.; Brahim, S.; de Waard, M.; Kenani, A. Doxorubicin coupled to penetratin promotes apoptosis in CHO cells by a mechanism involving c-Jun NH2-terminal kinase. Biochem. Biophys. Res. Commun. 2010, 396, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Tisseyre, C.; Ahmadi, M.; Bacot, S.; Dardevet, L.; Perret, P.; Ronjat, M.; Fagret, D.; Usson, Y.; Ghezzi, C.; de Waard, M. Quantitative evaluation of the cell penetrating properties of an iodinated Tyr-l-maurocalcine analog. Biochim. Biophys. Acta 2014, 1843, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk, K.; Ram, N.; Boisseau, S.; Strappazzon, F.; Rehaim, A.; Sadoul, R.; Darbon, H.; Ronjat, M.; de Waard, M. Critical amino acid residues of maurocalcine involved in pharmacology, lipid interaction and cell penetration. Biochim. Biophys. Acta 2007, 1768, 2528–2540. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Bacot, S.; Poillot, C.; Desruet, M.-D.; Perret, P.; Riou, L.; Cestèle, S.; Couvet, M.; Bourgoin, S.; Sève, M.; et al. Selective mono-radioiodination and characterization of a cell-penetrating peptide: l-Tyr-Maurocalcine. Radiochim. Acta 2014, 102, 1047–1057. [Google Scholar] [CrossRef]
- Maiti, N.R.; Surewicz, W.K. The role of disulfide bridge in the folding and stability of the recombinant human prion protein. J. Biol. Chem. 2001, 276, 2427–2431. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.S.; Povarnitsyna, T.V.; Glukhov, A.S.; Melnik, T.N.; Uversky, V.N.; Sarma, R.H. SS-Stabilizing proteins rationally: Intrinsic disorder-based design of stabilizing disulphide bridges in GFP. J. Biomol. Struct. Dyn. 2012, 29, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Friedman, A.M.; Rayudu, G.V.; Clark, P.; Fordham, E.W. In vivo stability and distribution of [131I]iodomethyl trimethylammonium chloride: Concise communication. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1980, 21, 679–681. [Google Scholar]
- Yu, K.O.; Narayanan, L.; Mattie, D.R.; Godfrey, R.J.; Todd, P.N.; Sterner, T.R.; Mahle, D.A.; Lumpkin, M.H.; Fisher, J.W. The pharmacokinetics of perchlorate and its effect on the hypothalamus–pituitary–thyroid axis in the male rat. Toxicol. Appl. Pharmacol. 2002, 182, 148–159. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perret, P.; Ahmadi, M.; Riou, L.; Bacot, S.; Pecher, J.; Poillot, C.; Broisat, A.; Ghezzi, C.; De Waard, M. Biodistribution, Stability, and Blood Distribution of the Cell Penetrating Peptide Maurocalcine in Mice. Int. J. Mol. Sci. 2015, 16, 27730-27740. https://doi.org/10.3390/ijms161126054
Perret P, Ahmadi M, Riou L, Bacot S, Pecher J, Poillot C, Broisat A, Ghezzi C, De Waard M. Biodistribution, Stability, and Blood Distribution of the Cell Penetrating Peptide Maurocalcine in Mice. International Journal of Molecular Sciences. 2015; 16(11):27730-27740. https://doi.org/10.3390/ijms161126054
Chicago/Turabian StylePerret, Pascale, Mitra Ahmadi, Laurent Riou, Sandrine Bacot, Julien Pecher, Cathy Poillot, Alexis Broisat, Catherine Ghezzi, and Michel De Waard. 2015. "Biodistribution, Stability, and Blood Distribution of the Cell Penetrating Peptide Maurocalcine in Mice" International Journal of Molecular Sciences 16, no. 11: 27730-27740. https://doi.org/10.3390/ijms161126054
APA StylePerret, P., Ahmadi, M., Riou, L., Bacot, S., Pecher, J., Poillot, C., Broisat, A., Ghezzi, C., & De Waard, M. (2015). Biodistribution, Stability, and Blood Distribution of the Cell Penetrating Peptide Maurocalcine in Mice. International Journal of Molecular Sciences, 16(11), 27730-27740. https://doi.org/10.3390/ijms161126054