
miR-218 Inhibits Erythroid Differentiation and Alters Iron Metabolism by Targeting ALAS2 in K562 Cells

 , ,
, ,
Abstract
:
1. Introduction
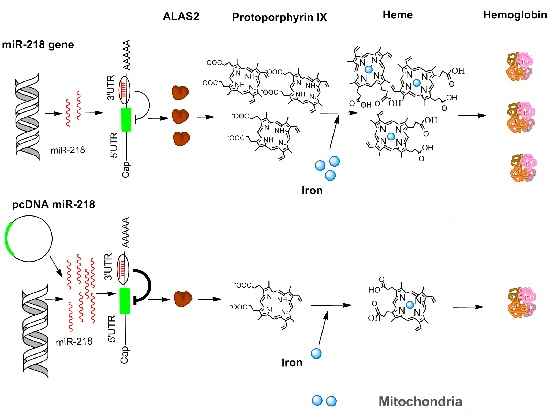
2. Results and Discussion
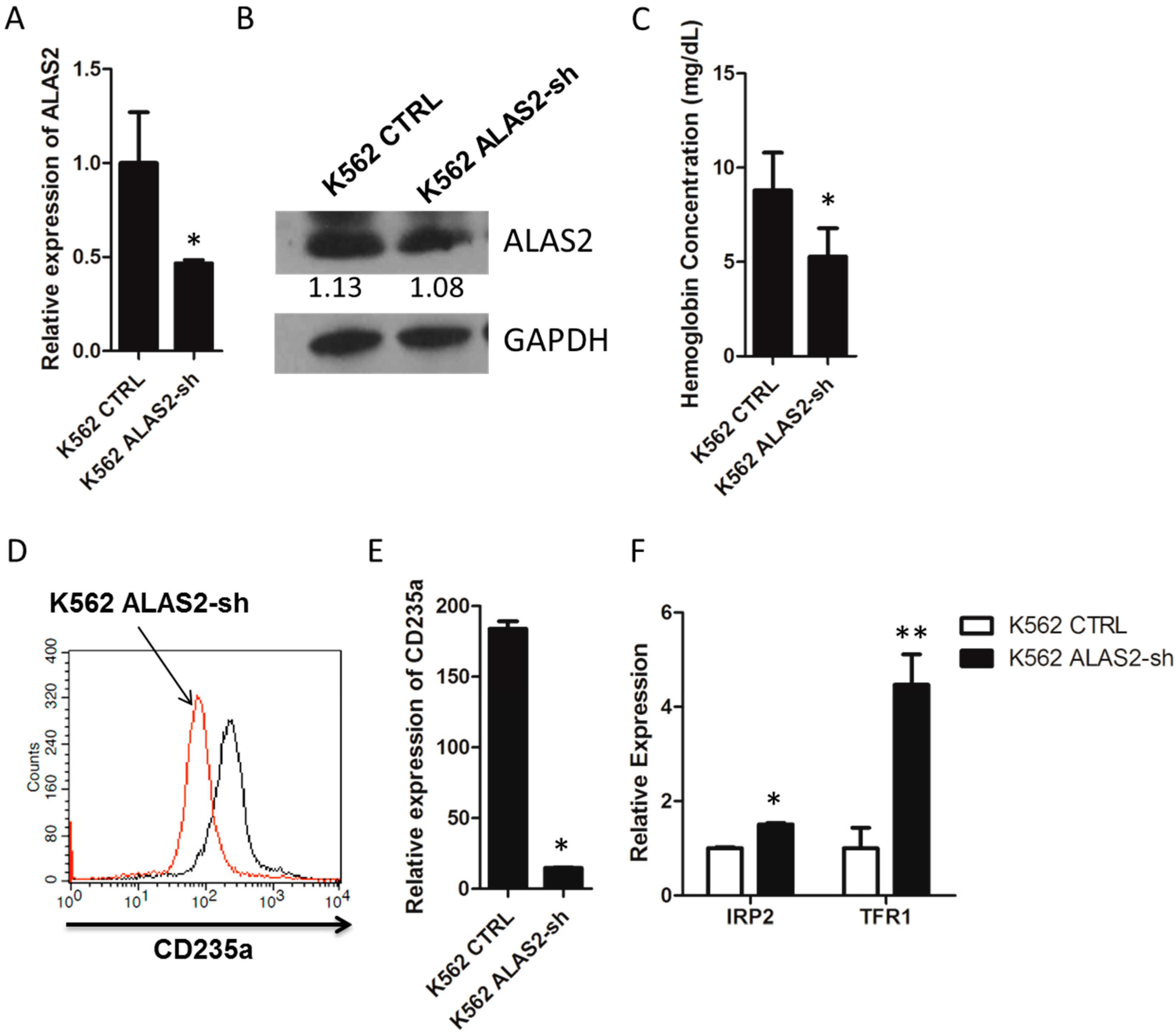
2.1. ALAS2 Knockdown Repressed Erythroid Gene Expression and Altered Iron Metabolism-Related Genes Expression in K562 Cells

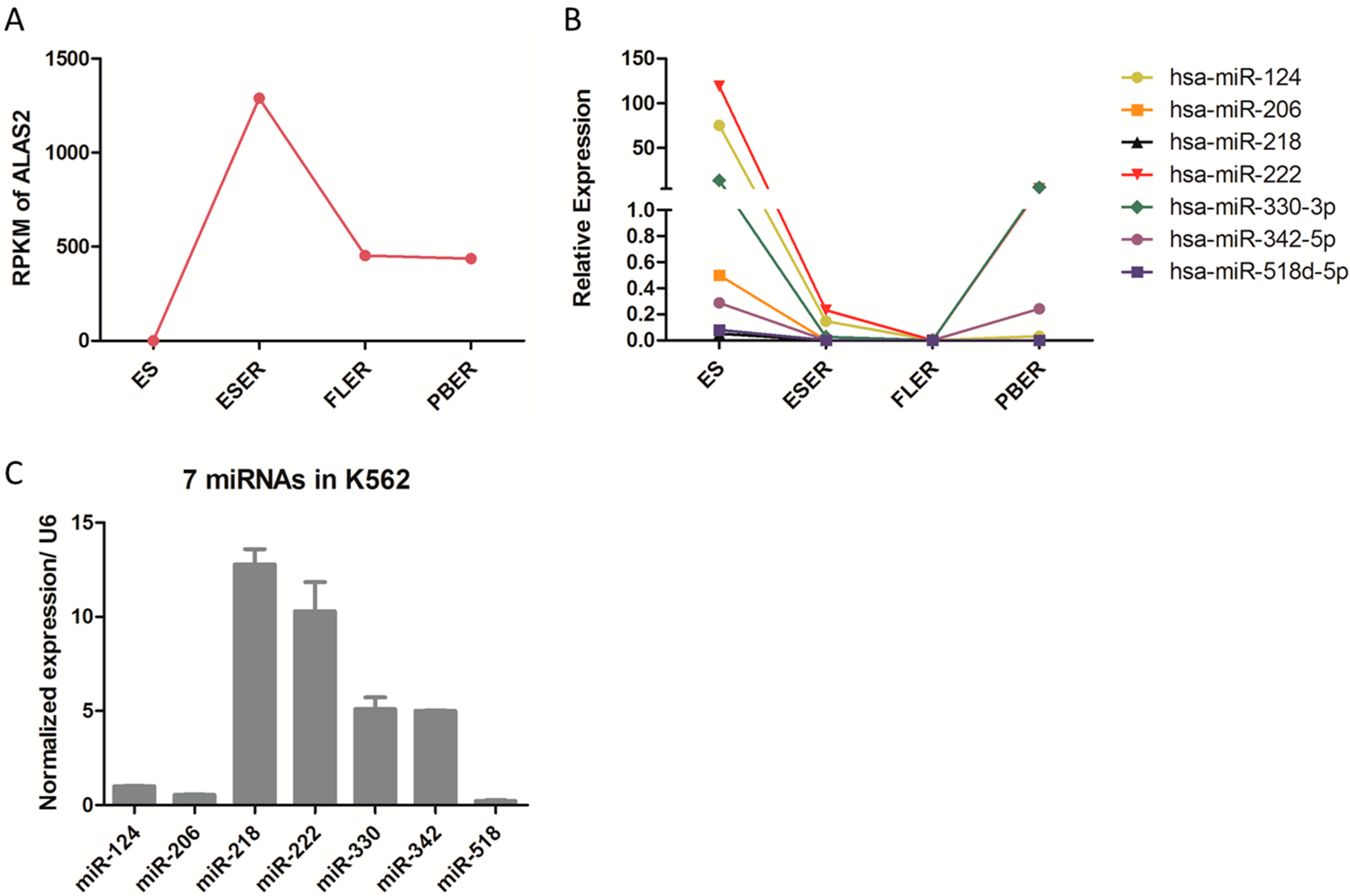
2.2. Candidate miRNAs Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | miRanda | microcosm | microrna.org | TargetScan |
|---|---|---|---|---|
| hsa-miR-1207-3p | - | - | - | √ |
| hsa-miR-124 | - | √ | - | - |
| hsa-miR-1254 | - | - | - | √ |
| hsa-miR-1257 | - | - | - | √ |
| hsa-miR-140 | √ | - | - | - |
| hsa-miR-142-5p | √ | √ | - | - |
| hsa-miR-202 | √ | √ | - | - |
| hsa-miR-206 | √ | - | - | - |
| hsa-miR-218 | √ | √ | √ | √ |
| hsa-miR-222 | √ | - | - | - |
| hsa-miR-24 | √ | √ | √ | - |
| hsa-miR-298 | - | √ | - | √ |
| hsa-miR-30a-3p | √ | - | - | - |
| hsa-miR-30e-3p | √ | - | - | - |
| hsa-miR-330 | √ | - | - | - |
| hsa-miR-330-3p | - | √ | - | √ |
| hsa-miR-342-5p | - | - | - | √ |
| hsa-miR-378 | - | √ | - | - |
| hsa-miR-422a | - | √ | - | - |
| hsa-miR-483-5p | - | √ | - | - |
| hsa-miR-500 | - | √ | - | - |
| hsa-miR-506 | √ | √ | - | - |
| hsa-miR-518d-5p | - | √ | - | - |
| hsa-miR-520f | √ | √ | - | - |
| hsa-miR-526a | √ | - | - | - |
| hsa-miR-565 | √ | √ | - | - |
| hsa-miR-571 | √ | √ | - | - |
| hsa-miR-608 | - | - | - | √ |
| hsa-miR-636 | - | - | - | √ |
| hsa-miR-661 | - | √ | - | - |
| hsa-miR-886-3p | - | √ | - | - |
| hsa-miR-891a | - | √ | - | - |
| hsa-miR-922 | - | √ | - | - |
| hsa-miR-92a-2* | - | √ | - | √ |
| hsa-miR-940 | - | - | - |


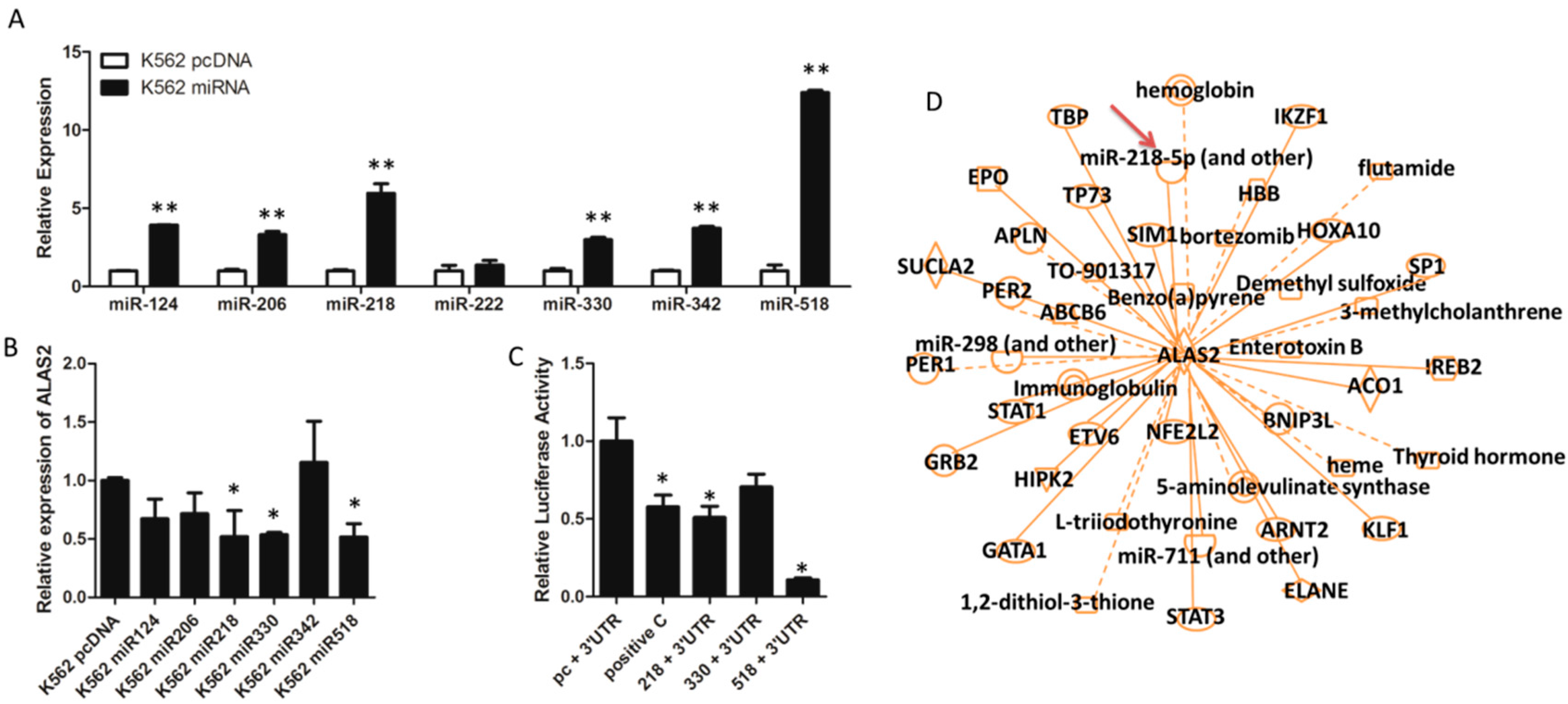
2.3. ALAS2 Is a Down-Regulated Target of miR-218

2.4. Iron Metabolism Is Regulated by miR-218 in K562 Cells

2.5. Overexpression of miR-218 Inhibits Induced Erythroid Differentiation in K562 Cells

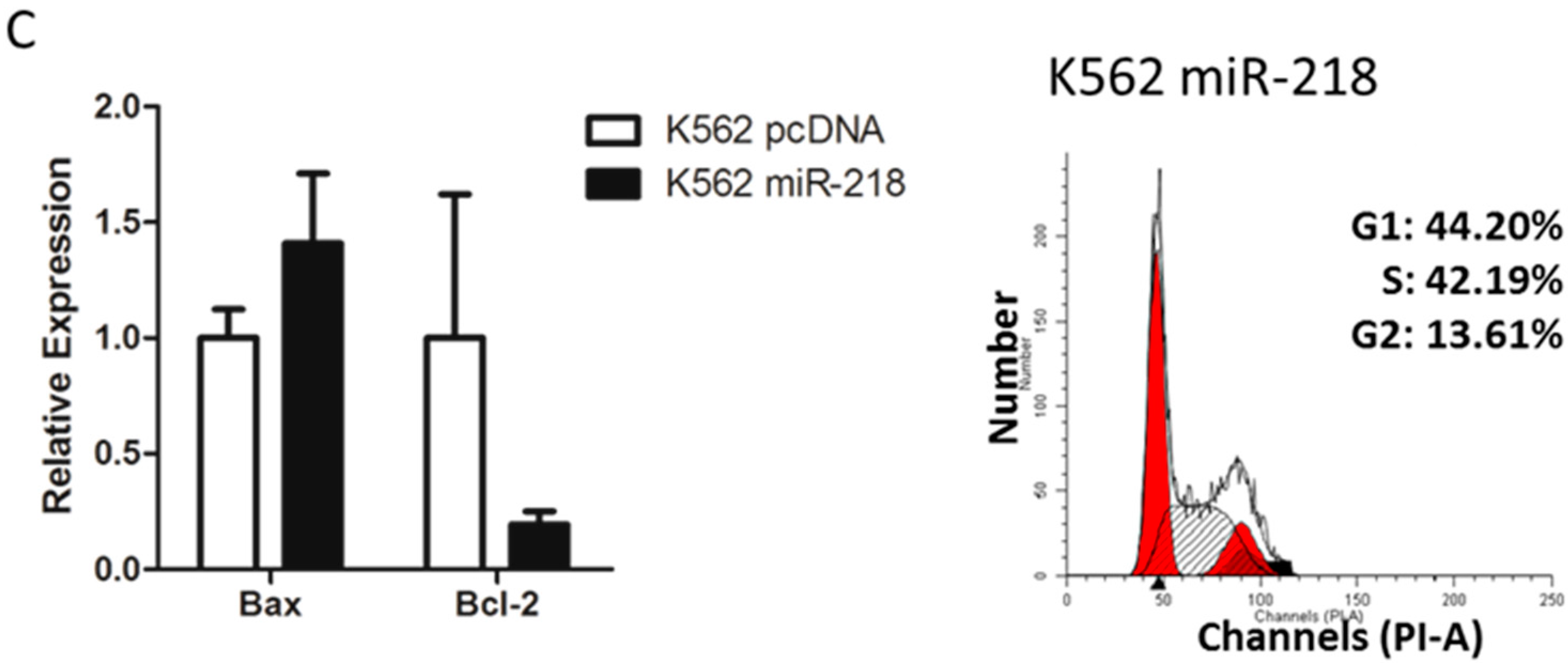
2.6. Hsa-miR-218 Plays a Role in the Proliferation and Apoptosis of K562 Cells


3. Experimental Section
3.1. Cell Culture
3.2. Transcriptome, miRNome Sequencing, and Bioinformatics Analyses
3.3. Plasmids Construction
3.4. Total RNA Extraction and Quantitative Real-Time PCR
3.5. Dual-Luciferase Reporter Assay
3.6. Mitochondria Isolation, Iron and Hemoglobin Quantification
3.7. Western Blot
3.8. Flow Cytometry
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, Q.; Huang, Z.; Xue, H.; Jin, C.; Ju, X.L.; Han, J.D.; Chen, Y.G. microRNA miR-24 inhibits erythropoiesis by targeting activin type I receptor ALK4. Blood 2008, 111, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Grabher, C.; Payne, E.M.; Johnston, A.B.; Bolli, N.; Lechman, E.; Dick, J.E.; Kanki, J.P.; Look, A.T. Zebrafish microRNA-126 determines hematopoietic cell fate through c-Myb. Leukemia 2011, 25, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Menne, T.F.; Scepanovic, D.; Vergilio, J.A.; Ji, P.; Kim, J.; Thiru, P.; Orkin, S.H.; Lander, E.S.; Lodish, H.F. microRNA-15a and -16–1 act via Myb to elevate fetal hemoglobin expression in human trisomy 13. Proc. Natl. Acad. Sci. USA 2011, 108, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Gabbianelli, M.; Testa, U.; Morsilli, O.; Pelosi, E.; Saulle, E.; Petrucci, E.; Castelli, G.; Giovinazzi, S.; Mariani, G.; Fiori, M.E.; et al. Mechanism of human hb switching: A possible role of the KIT receptor/miR 221–222 complex. Haematologica 2010, 95, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Pase, L.; Layton, J.E.; Kloosterman, W.P.; Carradice, D.; Waterhouse, P.M.; Lieschke, G.J. miR-451 regulates zebrafish erythroid maturation in vivo via its target GATA2. Blood 2009, 113, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Svasti, S.; Masaki, S.; Penglong, T.; Abe, Y.; Winichagoon, P.; Fucharoen, S.; Umemura, T. Expression of microRNA-451 in normal and thalassemic erythropoiesis. Ann. Hematol. 2010, 89, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Schreiber-Agus, N.; DePinho, R.A. Repression by the Mad(Mxi1)-Sin3 complex. Bioessays 1998, 20, 808–818. [Google Scholar] [CrossRef]
- Zhang, L.; Flygare, J.; Wong, P.; Lim, B.; Lodish, H.F. miR-191 regulates mouse erythroblast enucleation by down-regulating Riok3 and Mxi1. Genes Dev. 2011, 25, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Minotti, G.; Cairo, G. Iron regulatory proteins: From molecular mechanisms to drug development. Antioxid. Redox Signal. 2010, 13, 1593–1616. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.; Galy, B.; Schwanhaeusser, B.; Blake, J.; Bahr-Ivacevic, T.; Benes, V.; Selbach, M.; Muckenthaler, M.U.; Hentze, M.W. Iron regulatory protein-1 and -2: Transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins. Blood 2011, 118, e168–e179. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.; Gray, N.K.; Hentze, M.W. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol. Cell 1998, 2, 383–388. [Google Scholar] [CrossRef]
- Zhang, D.L.; Hughes, R.I.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Kobune, M.; Ohkubo, S.; Fujikawa, K.; Tanaka, M.; Takimoto, R.; Takada, K.; Takahari, D.; Kawano, Y.; Kohgo, Y.; et al. Iron/IRP-1-dependent regulation of mRNA expression for transferrin receptor, DMT1 and ferritin during human erythroid differentiation. Exp. Hematol. 2007, 35, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, I.; de Falco, L.; Asci, R.; Russo, R.; Colucci, S.; Gorrese, M.; Zollo, M.; Iolascon, A. Regulation of divalent metal transporter 1 (DMT1) non-IRE isoform by the microRNA let-7d in erythroid cells. Haematologica 2010, 95, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Chang, K.H.; Qu, H.; Zhang, Z.; Xiong, Q.; Qi, H.; Cui, P.; Lin, Q.; Ruan, X.; et al. Transcriptome dynamics during human erythroid differentiation and development. Genomics 2013, 102, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhu, Y.; Guo, L.; Dong, L.; Liu, H.; Yin, H.; Zhang, Z.; Li, Y.; Liu, C.; Ma, Y.; et al. A regulatory circuit comprising GATA1/2 switch and microRNA-27a/24 promotes erythropoiesis. Nucleic Acids Res. 2014, 42, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, O.; Iwasaki, S.; Hashimoto, Y. Hemin-induced erythroid differentiation of human myeloleukemia K562 cell line and its modification by bioresponse modifiers. Cell. Mol. Biol. 1997, 43, 115–134. [Google Scholar] [PubMed]
- Li, X.F.; Anderson, J.; Hutzler, D.; Roodman, G.D. Hemin-induced erythroid-differentiation changes the sensitivity of K562 cells to tumor necrosis factor-α. Exp. Hematol. 1989, 17, 1059–1062. [Google Scholar] [PubMed]
- Witt, O.; Sand, K.; Pekrun, A. Butyrate-induced erythroid differentiation of human K562 leukemia cells involves inhibition of ERK and activation of p38 MAP kinase pathways. Blood 2000, 95, 2391–2396. [Google Scholar] [PubMed]
- Bottomley, S.S.; May, B.K.; Cox, T.C.; Cotter, P.D.; Bishop, D.F. Molecular defects of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia. J. Bioenerg. Biomembr. 1995, 27, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Harigae, H.; Furuyama, K.; Kudo, K.; Hayashi, N.; Yamamoto, M.; Sassa, S.; Sasaki, T. A novel mutation of the erythroid-specific γ-aminolevulinate synthase gene in a patient with non-inherited pyridoxine-responsive sideroblastic anemia. Am. J. Hematol. 1999, 62, 112–114. [Google Scholar] [CrossRef]
- Harigae, H.; Furuyama, K.; Kimura, A.; Neriishi, K.; Tahara, N.; Kondo, M.; Hayashi, N.; Yamamoto, M.; Sassa, S.; Sasaki, T. A novel mutation of the erythroid-specific δ-aminolaevulinate synthase gene in a patient with X-linked sideroblastic anaemia. Br. J. Haematol. 1999, 106, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.D.; Richardson, D.R.; Prchal, J.; Ponka, P. Mitochondrial iron metabolism and sideroblastic anemia. Acta Haematol. 2009, 122, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Harigae, H.; Nakajima, O.; Suwabe, N.; Yokoyama, H.; Furuyama, K.; Sasaki, T.; Kaku, M.; Yamamoto, M.; Sassa, S. Aberrant iron accumulation and oxidized status of erythroid-specific δ-aminolevulinate synthase (ALAS2)-deficient definitive erythroblasts. Blood 2003, 101, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, O.; Takahashi, S.; Harigae, H.; Furuyama, K.; Hayashi, N.; Sassa, S.; Yamamoto, M. Heme deficiency in erythroid lineage causes differentiation arrest and cytoplasmic iron overload. EMBO J. 1999, 18, 6282–6289. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Kozaki, K.I.; Tanaka, S.; Arii, S.; Imoto, I.; Inazawa, J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis 2010, 31, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Fang, X. A brief review on the human encyclopedia of DNA elements (encode) project. Genom. Proteom. Bioinform. 2013, 11, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Yang, Y.; Beijing Institute of Genomics, Chinese Academy of Sciences. Personal communication, 2013.
- Friedlander, M.R.; Chen, W.; Adamidi, C.; Maaskola, J.; Einspanier, R.; Knespel, S.; Rajewsky, N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008, 26, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Zhang, Z.; Chang, K.H.; Qu, H.; Wang, H.; Qi, H.; Li, Y.; Ruan, X.; Yang, Y.; Sandstrom, R.; et al. Comprehensive characterization of erythroid-specific enhancers in the genomic regions of human Kruppel-like factors. BMC Genom. 2013, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Yang, B.; Yang, J.; Ruan, X.; Yang, Y.; Wakeland, E.K.; Li, Q.; Fang, X. Influence of carbon monoxide on growth and apoptosis of human umbilical artery smooth muscle cells and vein endothelial cells. Int. J. Biol. Sci. 2012, 8, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Liu, S.; Sun, H.; Yang, Y.; Qi, H.; Ding, N.; Zheng, J.; Dong, X.; Qu, H.; Zhang, Z.; et al. miR-218 Inhibits Erythroid Differentiation and Alters Iron Metabolism by Targeting ALAS2 in K562 Cells. Int. J. Mol. Sci. 2015, 16, 28156-28168. https://doi.org/10.3390/ijms161226088
Li Y, Liu S, Sun H, Yang Y, Qi H, Ding N, Zheng J, Dong X, Qu H, Zhang Z, et al. miR-218 Inhibits Erythroid Differentiation and Alters Iron Metabolism by Targeting ALAS2 in K562 Cells. International Journal of Molecular Sciences. 2015; 16(12):28156-28168. https://doi.org/10.3390/ijms161226088
Chicago/Turabian StyleLi, Yanming, Shuge Liu, Hongying Sun, Yadong Yang, Heyuan Qi, Nan Ding, Jiawen Zheng, Xunong Dong, Hongzhu Qu, Zhaojun Zhang, and et al. 2015. "miR-218 Inhibits Erythroid Differentiation and Alters Iron Metabolism by Targeting ALAS2 in K562 Cells" International Journal of Molecular Sciences 16, no. 12: 28156-28168. https://doi.org/10.3390/ijms161226088
APA StyleLi, Y., Liu, S., Sun, H., Yang, Y., Qi, H., Ding, N., Zheng, J., Dong, X., Qu, H., Zhang, Z., & Fang, X. (2015). miR-218 Inhibits Erythroid Differentiation and Alters Iron Metabolism by Targeting ALAS2 in K562 Cells. International Journal of Molecular Sciences, 16(12), 28156-28168. https://doi.org/10.3390/ijms161226088