
A Time to Kill: Targeting Apoptosis in Cancer

{kind=link}
Abstract
:1. Introduction
2. Pathways to Apoptosis

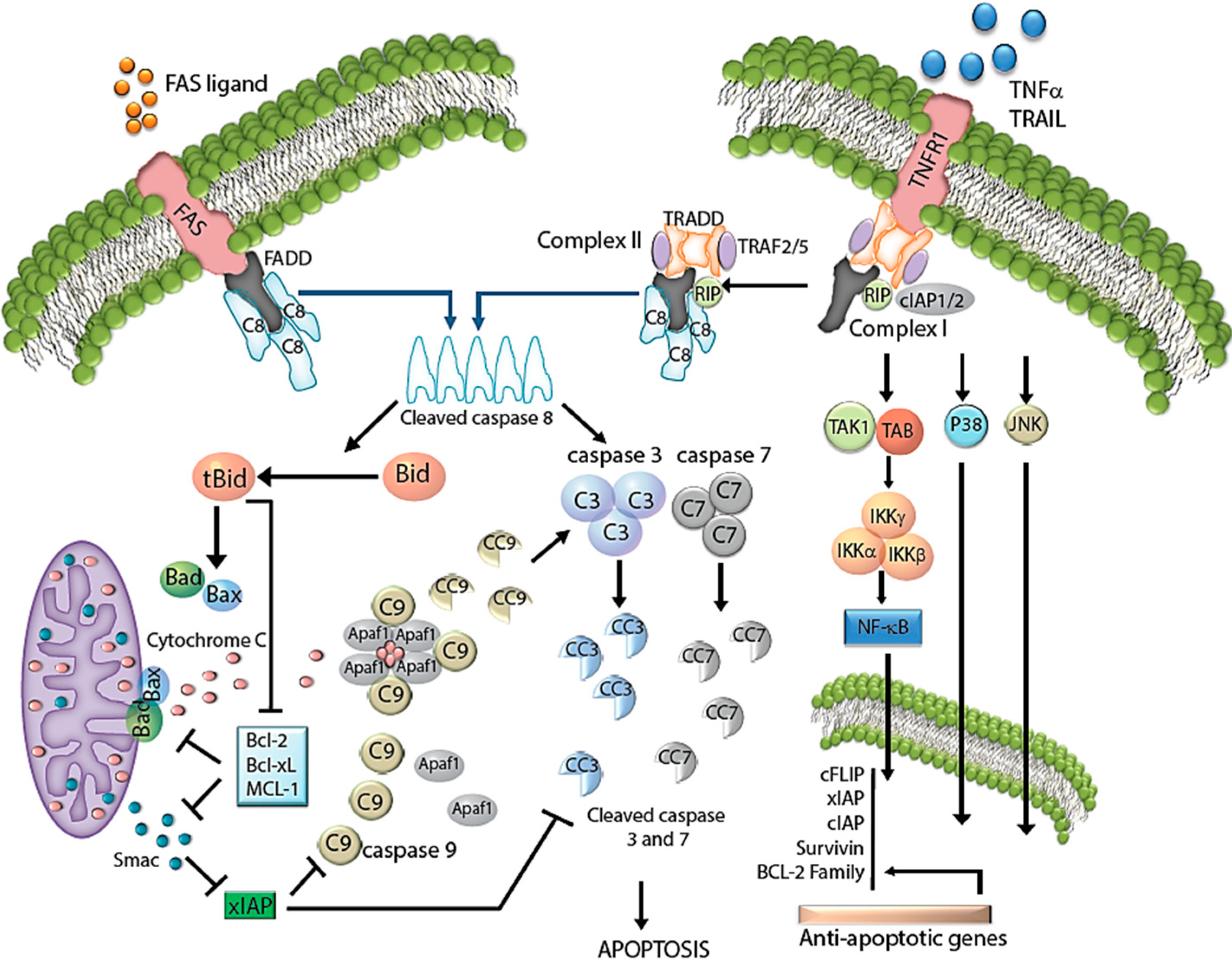
2.1. Extrinsic Pathway
2.2. Regulation of the Extrinsic Pathway
2.3. Intrinsic Pathway
2.4. Regulation of the Intrinsic Pathway
2.5. Lysosomal Mitochondrial Pathway
3. Evasion of Apoptosis by Cancer Cells
3.1. Disruption of the First Apoptotic Signal (FAS) and DR5 Response
3.2. Disruption in the Balance of Bcl2 Proteins
3.3. Dysregulation of Inhibitor of Apoptosis (IAPs)
3.4. Reduced Caspase Activity
4. Targeting Apoptosis Pathways in Cancer Treatment
4.1. TNF-Related Apoptosis-Inducing Ligand (TRAIL)
4.2. Bcl2 Family
4.3. IAP Inhibitors
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tomei, L.D.; Cope, F.O. Apoptosis: The Molecular Basis of Cell Death; Books on Demand: Stoughton, WI, USA, 1991. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell. Death Differ. 2008, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Robbins, S.L.; Kumar, V.; Abbas, A.K.; Cotran, R.S.; Fausto, N. Robbins and Cotran’s Pathologic Basis of Disease; Elsevier Health Sciences: Makati City, Philippines, 2009. [Google Scholar]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.-G. Cleavage of the death domain kinase RIP by Caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Long, J.S.; Ryan, K.M. New frontiers in promoting tumour cell death: Targeting apoptosis, necroptosis and autophagy. Oncogene 2012, 31, 5045–5060. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Dixit, V.M. Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. 2009, 17, 14–24. [Google Scholar] [CrossRef]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Dewson, G.; Kratina, T.; Czabotar, P.; Day, C.L.; Adams, J.M.; Kluck, R.M. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol. Cell. 2009, 36, 696–703. [Google Scholar] [CrossRef]
- Goldstein, J.C.; Waterhouse, N.J.; Juin, P.; Evan, G.I.; Green, D.R. The coordinate release of cytochrome C during apoptosis is rapid, complete and kinetically invariant. Nat. Cell. Biol. 2000, 2, 156–162. [Google Scholar] [PubMed]
- Wei, M.C.; Zong, W.-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1–caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondrial Regulation of Cell Death. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, E.C.; Mahoney, D.J.; Cheung, H.H.; Plenchette, S.; Baird, S.; Korneluk, R.G. IAP-targeted therapies for cancer. Oncogene 2008, 27, 6252–6275. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Meier, P. Inhibitor of apoptosis (IAP) proteins—Modulators of cell death and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Estornes, Y.; Bertrand, M.J.M. IAPs, regulators of innate immunity and inflammation. Semin. Cell Dev. Biol. 2014. [Google Scholar] [CrossRef]
- Chen, D.J.; Huerta, S. Smac mimetics as new cancer therapeutics. Anticancer Drugs 2009, 20, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Suh, W.-K.; Jin, J.; Woo, M.; Du, C.; Elia, A.; Duncan, G.S.; Wakeham, A.; Itie, A.; Lowe, S.W.; et al. Generation and characterization of smac/DIABLO-deficient mice. Mol. Cell. Biol. 2002, 22, 3509–3517. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Datta, P.; Srinivasula, S.M.; Ji, W.; Gupta, S.; Zhang, Z.; Davies, E.; Hajnoczky, G.; Saunders, T.L.; van Keuren, M.L.; et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of MND2 mutant mice. Nature 2003, 425, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.M.; Morrison, A.; Klupsch, K.; Fedele, V.; Moisoi, N.; Teismann, P.; Abuin, A.; Grau, E.; Geppert, M.; Livi, G.P.; et al. Neuroprotective role of the reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell. Biol. 2004, 24, 9848–9862. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Guan, S.; Wang, H.; Burlingame Alma, L.; Wells James, A. Substrates of IAP ubiquitin ligases identified with a designed orthogonal E3 ligase, the NEDDylator. Mol. Cell 2013, 49, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in beclin-1. 2007, 26, 1527–2539. [Google Scholar]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.-U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell. Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Foghsgaard, L.; Wissing, D.; Mauch, D.; Lademann, U.; Bastholm, L.; Boes, M.; Elling, F.; Leist, M.; Jäättelä, M. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 2001, 153, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: from waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.-C.; Appelqvist, H.; Nilsrighson, C.; Kågedal, K.; Roberg, K.; Öllinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, N.; Chen, T.; Han, Y.; Li, C.; Wang, Y.; He, W.; Zhang, L.; Wan, T.; Cao, X. The lysosome-associated apoptosis-inducing protein containing the pleckstrin homology (PH) and FYVE domains (LAPF), representative of a novel family of PH and FYVE domain-containing proteins, induces caspase-independent apoptosis via the lysosomal-mitochondrial pathway. J. Biol. Chem. 2005, 280, 40985–40995. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Hashemi, M.; Ande, S.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.; Kadkhoda, K.; Wiechec, E.; Halayko, A.; et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Jager, U.; Bocskor, S.; Le, T.; Mitterbauer, G.; Bolz, I.; Chott, A.; Kneba, M.; Mannhalter, C.; Nadel, B. Follicular lymphomas’ BCL-2/IgH junctions contain templated nucleotide insertions: Novel insights into the mechanism of t(14;18) translocation. Blood 2000, 95, 3520–3529. [Google Scholar] [PubMed]
- Adachi, M.; Tefferi, A.; Greipp, P.R.; Kipps, T.J.; Tsujimoto, Y. Preferential linkage of bcl-2 to immunoglobulin light chain gene in chronic lymphocytic leukemia. J. Exp. Med. 1990, 171, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Sanger, W.G.; Horsman, D.E.; Rosenwald, A.; Pickering, D.L.; Dave, B.; Dave, S.; Xiao, L.; Cao, K.; Zhu, Q.; et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol. 2004, 165, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Roulland, S.; Navarro, J.-M.; Grenot, P.; Milili, M.; Agopian, J.; Montpellier, B.; Gauduchon, P.; Lebailly, P.; Schiff, C.; Nadel, B. Follicular lymphoma-like B cells in healthy individuals: A novel intermediate step in early lymphomagenesis. J. Exp. Med. 2006, 203, 2425–2431. [Google Scholar] [CrossRef] [PubMed]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Nam, S.W.; Kim, C.J.; Cho, Y.G.; Lee, J.H.; Kim, H.S.; Park, W.S.; Kim, S.H.; et al. Inactivating mutations of proapoptotic Bad gene in human colon cancers. Carcinogenesis 2004, 25, 1371–1376. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Mesri, M.; Wall, N.R.; Li, J.; Kim, R.W.; Altieri, D.C. Cancer gene therapy using a survivin mutant adenovirus. J. Clin. Investig. 2001, 108, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Byun, D.-S.; Cho, K.; Ryu, B.-K.; Lee, M.-G.; Kang, M.-J.; Kim, H.-R.; Chi, S.-G. Hypermethylation of XIAP-associated factor 1, a putative tumor suppressor gene from the 17p13.2 locus, in human gastric adenocarcinomas. Cancer Res. 2003, 63, 7068–7075. [Google Scholar] [PubMed]
- Yang, D.; Welm, A.; Bishop, J.M. Cell division and cell survival in the absence of survivin. Proc. Natl. Acad. Sci. USA 2004, 101, 15100–15105. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, L.; Lovly, C.M.; Ratner, L. The role of NF-κB-1 and NF-κB-2-mediated resistance to apoptosis in lymphomas. Proc. Natl. Acad. Sci. USA 2006, 103, 9220–9225. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Debatin, K.-M.; Krammer, P.H. Death receptors in chemotherapy and cancer. Oncogene 2004, 23, 2950–2966. [Google Scholar] [CrossRef] [PubMed]
- Özören, N.; El-Deiry, W.S. Cell surface death receptor signaling in normal and cancer cells. Semin. Cancer Biol. 2003, 13, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Mérino, D.; Khaw, S.L.; Glaser, S.P.; Anderson, D.J.; Belmont, L.D.; Wong, C.; Yue, P.; Robati, M.; Phipson, B.; Fairlie, W.D.; et al. Bcl-2, Bcl-xL, and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood 2012, 119, 5807–5816. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.S.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Malek, M.; Zha, J.; Yue, P.; Kassees, R.; Berry, L.; Fairbrother, W.J.; Sampath, D.; Belmont, L.D. Navitoclax enhances the efficacy of taxanes in non–small cell lung cancer models. Clin. Cancer Res. 2011, 17, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Cragg, M.S.; Harris, C.; Strasser, A.; Scott, C.L. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat. Rev. Cancer 2009, 9, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, C.J.; Cory, S. ABT-199, a new Bcl-2–specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood 2013, 121, 2285–2288. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, F.; Merino, D.; Lee, L.; Breslin, K.; Pal, B.; Ritchie Matthew, E.; Smyth Gordon, K.; Christie, M.; Phillipson Louisa, J.; Burns Christopher, J.; et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell 2013, 24, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Touzeau, C.; Dousset, C.; le Gouill, S.; Sampath, D.; Leverson, J.D.; Souers, A.J.; Maiga, S.; Bene, M.C.; Moreau, P.; Pellat-Deceunynck, C.; et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia 2014, 28, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.C.S.; Seymour, J.F.; Bell, A.; Westerman, D.A.; Juneja, S.; Huang, D.C.S.; Roberts, A.W. Selective Bcl-2 inhibition with ABT-199 is highly active against chronic lymphocytic leukemia (CLL) irrespective of TP53 mutation or dysfunction. Blood 2013, 122, 1304–1304. [Google Scholar]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, R.; Fulda, S.; Carosati, E. IAP antagonists: Promising candidates for cancer therapy. Drug Discov. Today 2010, 15, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Neri, P.; Velankar, M.; Podar, K.; Hideshima, T.; Fulciniti, M.; Tassone, P.; Raje, N.; Mitsiades, C.; Mitsiades, N.; et al. Targeting mitochondrial factor Smac/DIABLO as therapy for multiple myeloma (MM). Blood 2007, 109, 1220–1227. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weisberg, E.; Kung, A.L.; Wright, R.D.; Moreno, D.; Catley, L.; Ray, A.; Zawel, L.; Tran, M.; Cools, J.; Gilliland, G.; et al. Potentiation of antileukemic therapies by Smac mimetic, LBW242: effects on mutant FLT3-expressing cells. Mol. Cancer Ther. 2007, 6, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, L.; Belz, K.; Fulda, S. Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Belz, K.; Schoeneberger, H.; Wehner, S.; Weigert, A.; Bonig, H.; Klingebiel, T.; Fichtner, I.; Fulda, S. Smac mimetic and glucocorticoids synergize to induce apoptosis in childhood ALL by promoting ripoptosome assembly. Blood 2014, 124, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; McEachern, D.; Li, W.; Davis, M.A.; Li, H.; Morgan, M.A.; Bai, L.; Sebolt, J.T.; Sun, H.; Lawrence, T.S.; et al. Radiosensitization of head and neck squamous cell carcinoma by a SMAC-mimetic compound, SM-164, requires activation of caspases. Mol. Cancer Ther. 2011, 10, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, G.; Zhao, Y.; Liu, G.; Wang, Y.; Ma, X.; Li, D.; Wu, Y.; Lu, J. Smac mimetic SM-164 potentiates APO2L/TRAIL- and doxorubicin-mediated anticancer activity in human hepatocellular carcinoma cells. PLoS One 2012, 7, e51461. [Google Scholar] [CrossRef] [PubMed]
- Ramachandiran, S.; Cain, J.; Liao, A.; He, Y.; Guo, X.; Boise, L.H.; Fu, H.; Ratner, L.; Khoury, H.J.; Bernal-Mizrachi, L. The Smac mimetic RMT5265.2HCL induces apoptosis in EBV and HTLV-I associated lymphoma cells by inhibiting XIAP and promoting the mitochondrial release of cytochrome C and Smac. Leuk. Res. 2012, 36, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Sheng, R.; Sun, H.; Liu, L.; Lu, J.; McEachern, D.; Wang, G.; Wen, J.; Min, P.; Du, Z.; Lu, H.; et al. A potent bivalent Smac mimetic (SM-1200) achieving rapid, complete, and durable tumor regression in mice. J. Med. Chem. 2013, 56, 3969–3979. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koff, J.L.; Ramachandiran, S.; Bernal-Mizrachi, L. A Time to Kill: Targeting Apoptosis in Cancer. Int. J. Mol. Sci. 2015, 16, 2942-2955. https://doi.org/10.3390/ijms16022942
Koff JL, Ramachandiran S, Bernal-Mizrachi L. A Time to Kill: Targeting Apoptosis in Cancer. International Journal of Molecular Sciences. 2015; 16(2):2942-2955. https://doi.org/10.3390/ijms16022942
Chicago/Turabian StyleKoff, Jean L., Sampath Ramachandiran, and Leon Bernal-Mizrachi. 2015. "A Time to Kill: Targeting Apoptosis in Cancer" International Journal of Molecular Sciences 16, no. 2: 2942-2955. https://doi.org/10.3390/ijms16022942
APA StyleKoff, J. L., Ramachandiran, S., & Bernal-Mizrachi, L. (2015). A Time to Kill: Targeting Apoptosis in Cancer. International Journal of Molecular Sciences, 16(2), 2942-2955. https://doi.org/10.3390/ijms16022942