
Methemoglobin Is an Endogenous Toll-Like Receptor 4 Ligand—Relevance to Subarachnoid Hemorrhage

Abstract
:1. Introduction
2. Results and Discussion
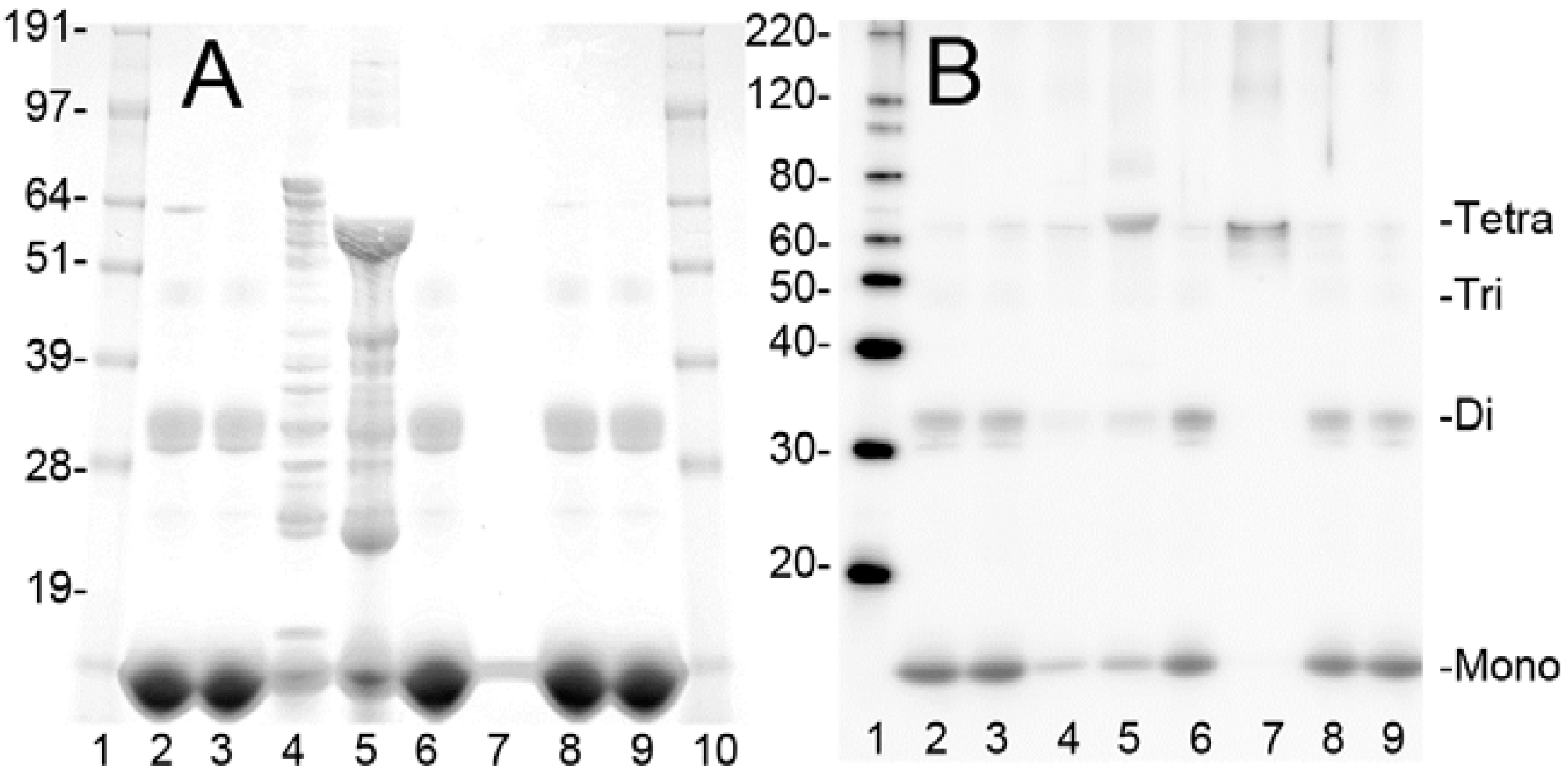
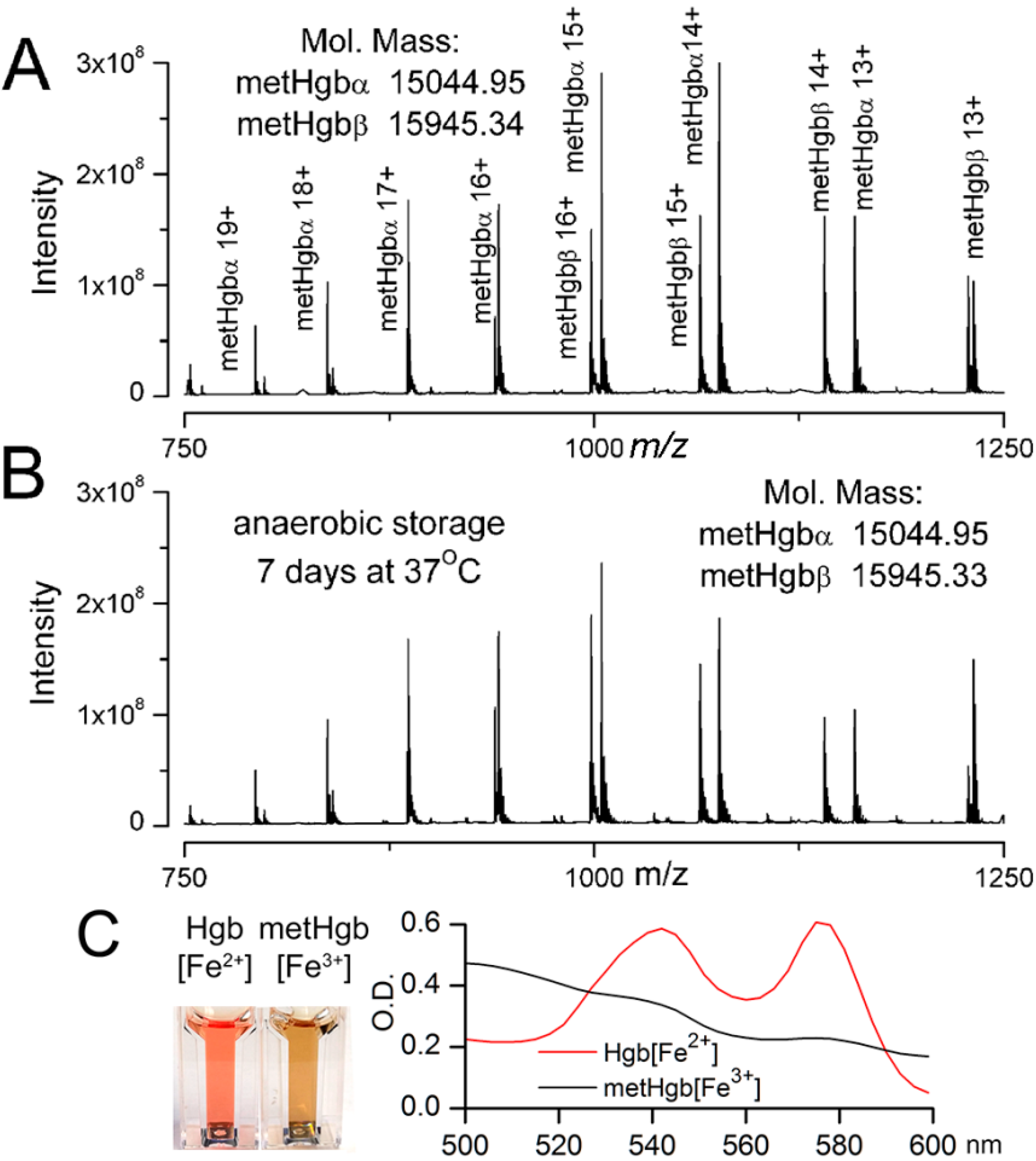
2.1. Purification of Bovine metHgb

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Steps | Protein (mg) | Yield (%) |
|---|---|---|
| Starting hemolysate | 2094 | 100 |
| Centrifugation | 2017 | 96 |
| Q-Sepharose | 1822 | 87 |
| Ultrafiltration | 1651 | 79 |
| PD-10 | 1374 | 66 |
| EndoTrap HD | 1364 | 65 |


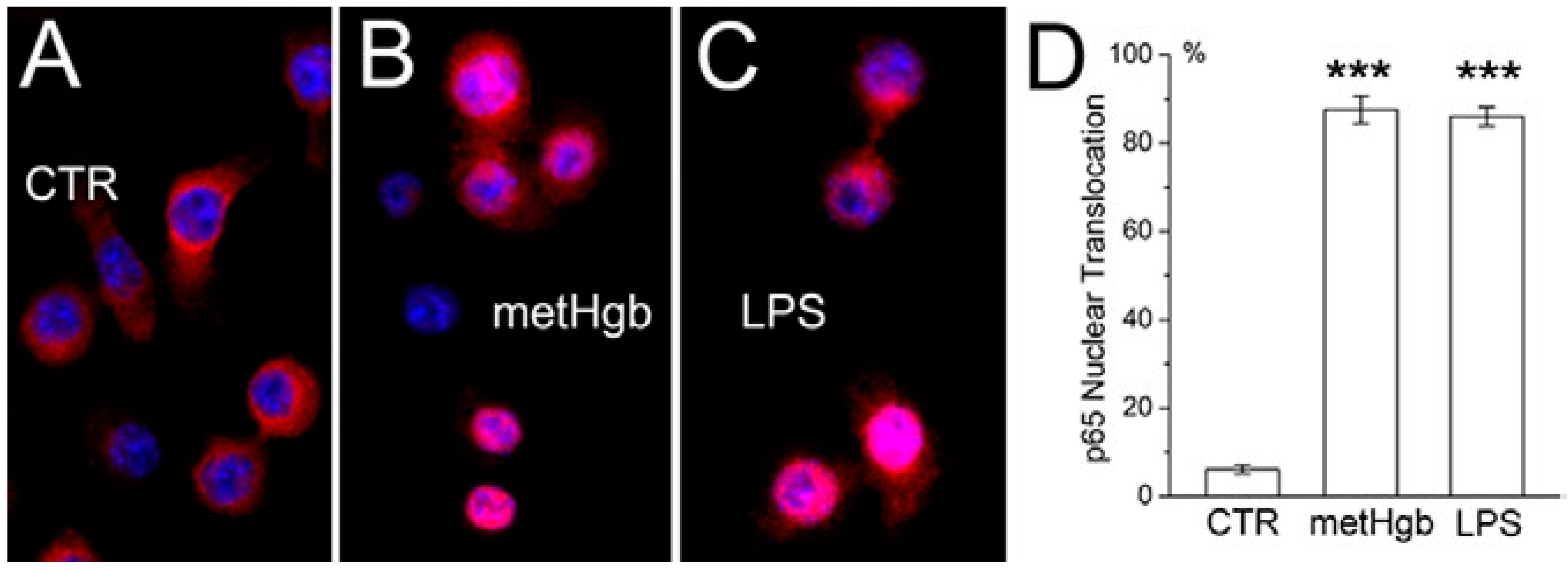
2.2. metHgb Induces p65 Nuclear Translocation in Microglia

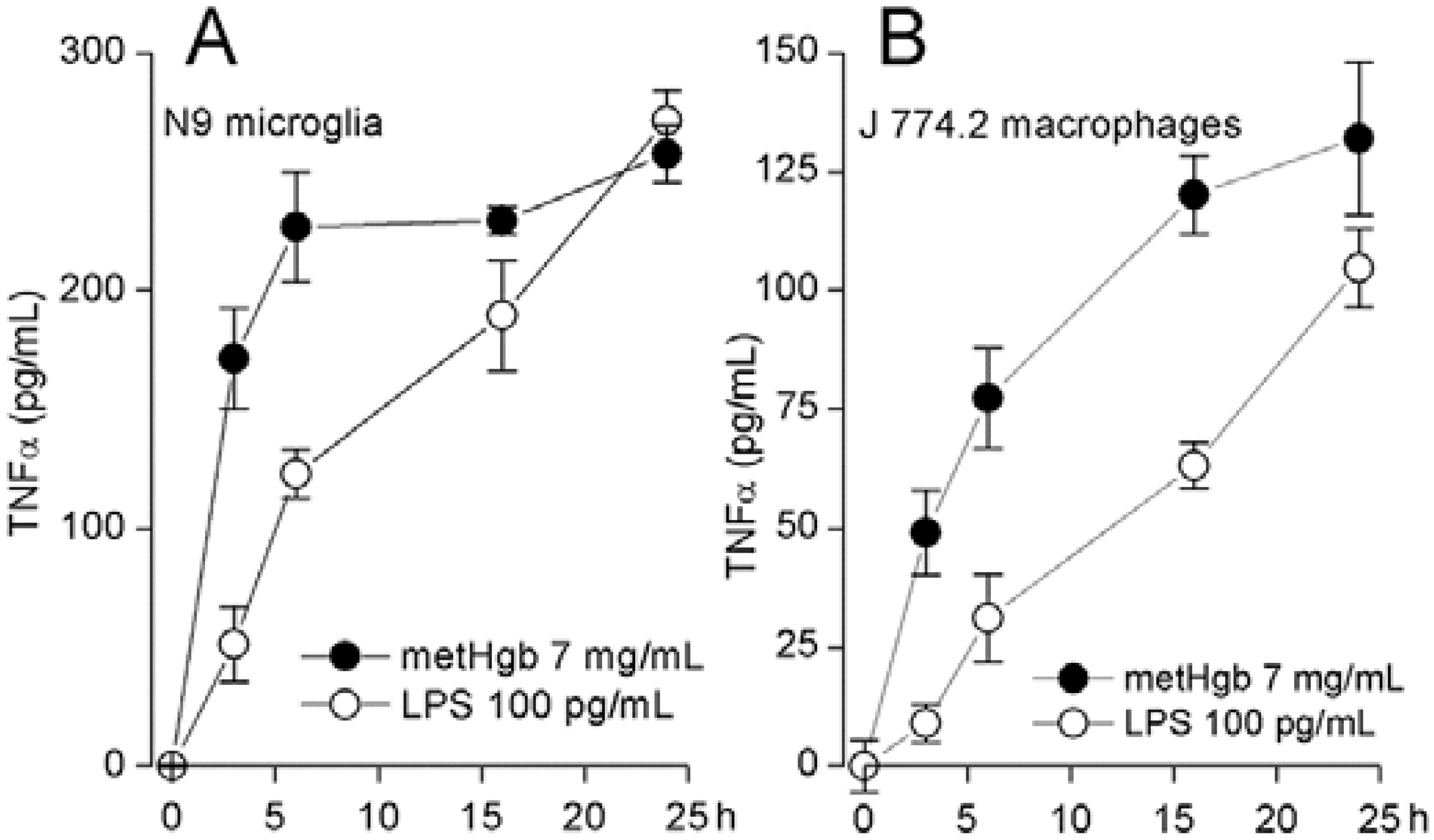
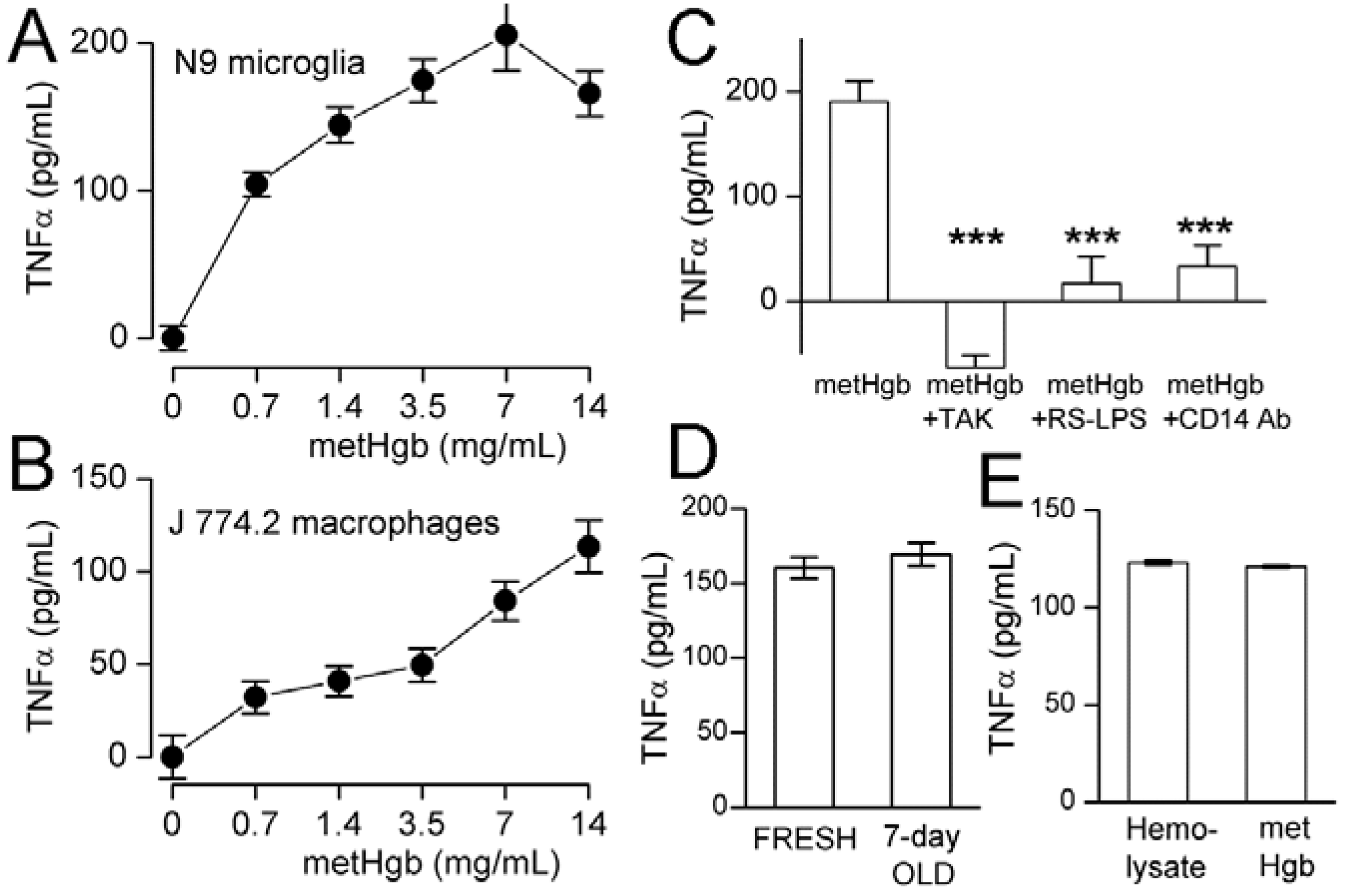
2.3. metHgb Induces Secretion of TNFα in Microglia and Macrophages

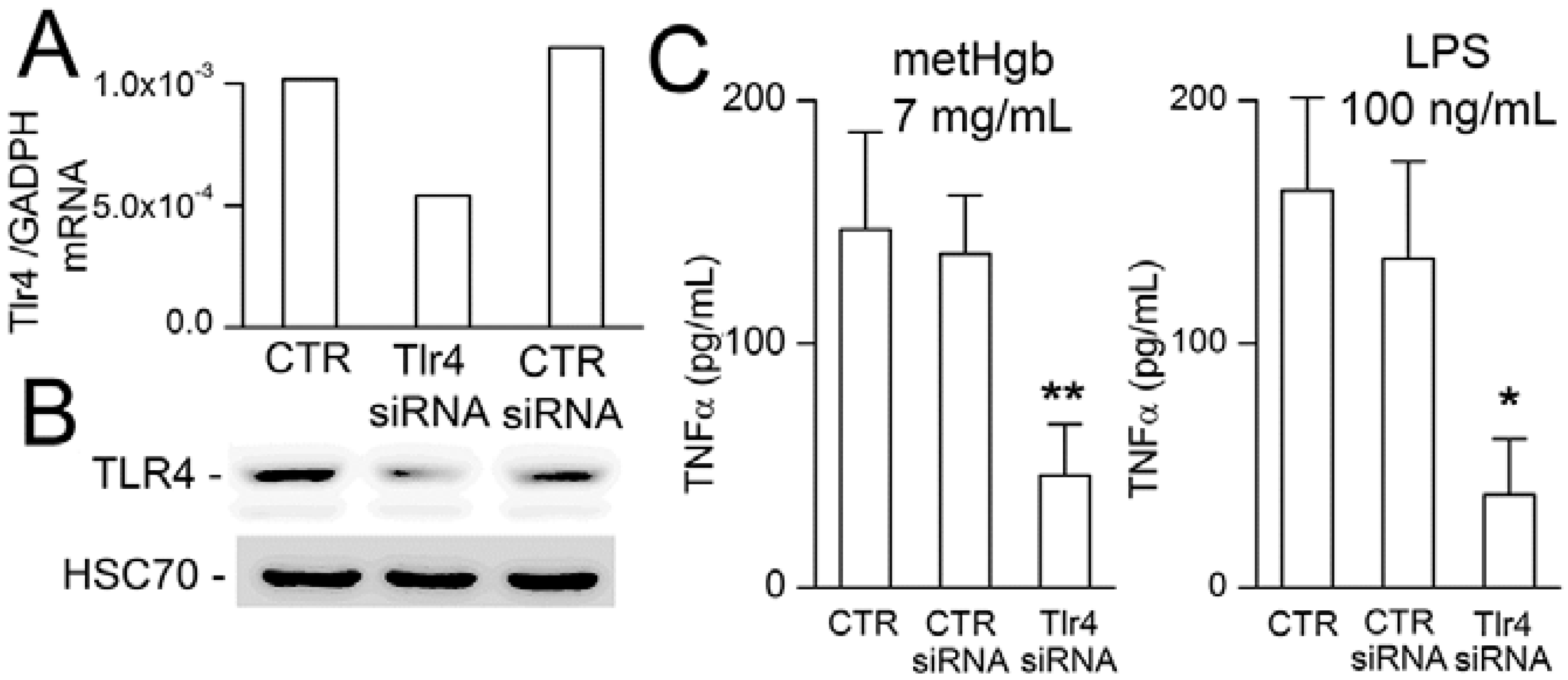
2.4. metHgb-Induced TNFα Secretion Is TLR4-Dependent


2.5. metHgb Is the Main Constituent of Hemolysate Responsible for TLR4 Activation
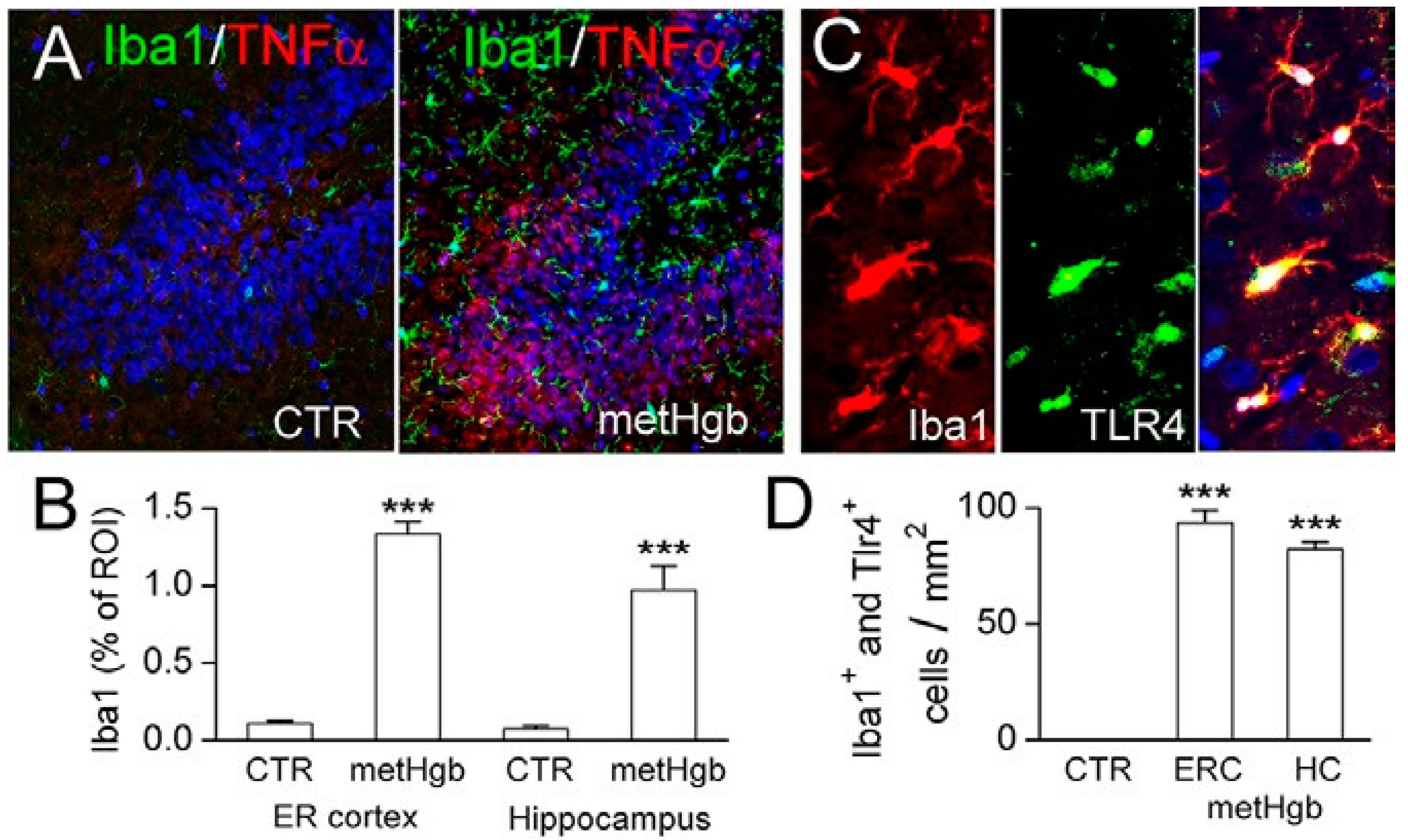
2.6. metHgb Induces Neuroinflammation

2.7. Discussion
3. Experimental Section
3.1. Purification of Bovine metHgb
3.2. Mass Spectrometry
3.3. Cell Culture
3.4. TNFα ELISA
3.5. siRNA Transfection
3.6. In Vivo metHgb Infusion
3.7. Immunohistochemistry
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2014, 10, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Ogden, J.A.; Utley, T.; Mee, E.W. Neurological and psychosocial outcome 4 to 7 years after subarachnoid hemorrhage. Neurosurgery 1997, 41, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Hackett, M.L.; Anderson, C.S. Health outcomes 1 year after subarachnoid hemorrhage: An international population-based study. The Australian Cooperative Research on Subarachnoid Hemorrhage Study Group. Neurology 2000, 55, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, K.T.; Copeland, D.; Bernardini, G.L.; Bates, J.E.; Peery, S.; Claassen, J.; Du, Y.E.; Stern, Y.; Connolly, E.S.; Mayer, S.A. Predictors of cognitive dysfunction after subarachnoid hemorrhage. Stroke 2002, 33, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Al-Khindi, T.; Macdonald, R.L.; Schweizer, T.A. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 2010, 41, e519–e536. [Google Scholar] [CrossRef] [PubMed]
- Haley, E.C., Jr. Measuring cognitive outcome after subarachnoid hemorrhage. Ann. Neurol. 2006, 60, 502–504. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.K.; Ilodigwe, D.; Li, Z.; Schweizer, T.A.; Macdonald, R.L. Global cerebral atrophy after subarachnoid hemorrhage: A possible marker of acute brain injury and assessment of its impact on outcome. Acta. Neurochir. Suppl. 2013, 115, 17–21. [Google Scholar] [PubMed]
- Bendel, P.; Koivisto, T.; Niskanen, E.; Kononen, M.; Aikia, M.; Hanninen, T.; Koskenkorva, P.; Vanninen, R. Brain atrophy and neuropsychological outcome after treatment of ruptured anterior cerebral artery aneurysms: A voxel-based morphometric study. Neuroradiology 2009, 51, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.K.; Kapadia, A.; Ilodigwe, D.; Li, Z.; Schweizer, T.A.; Macdonald, R.L. Impact of global cerebral atrophy on clinical outcome after subarachnoid hemorrhage. J. Neurosurg. 2013, 119, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Borges, I.T.; Shea, C.D.; Ohayon, J.; Jones, B.C.; Stone, R.D.; Ostuni, J.; Shiee, N.; McFarland, H.; Bielekova, B.; Reich, D.S. The effect of daclizumab on brain atrophy in relapsing-remitting multiple sclerosis. Mult. Scler. Relat. Disord. 2013, 2, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Novak, V.; Zhao, P.; Manor, B.; Sejdic, E.; Alsop, D.; Abduljalil, A.; Roberson, P.K.; Munshi, M.; Novak, P. Adhesion molecules, altered vasoreactivity, and brain atrophy in type 2 diabetes. Diabetes Care 2011, 34, 2438–2441. [Google Scholar] [CrossRef] [PubMed]
- Rocca, M.A.; Mondria, T.; Valsasina, P.; Sormani, M.P.; Flach, Z.H.; Te Boekhorst, P.A.; Comi, G.; Hintzen, R.Q.; Filippi, M. A three-year study of brain atrophy after autologous hematopoietic stem cell transplantation in rapidly evolving secondary progressive multiple sclerosis. Am. J. Neuroradiol. 2007, 28, 1659–1661. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.M.; Fox, N.C.; Miller, D.H. Magnetic resonance imaging measures of brain atrophy in multiple sclerosis. J. Magn. Reson. Imaging 2006, 23, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C. Endogenous ligands of TLR2 and TLR4: Agonists or assistants? J. Leukoc. Biol. 2010, 87, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Hauss-Wegrzyniak, B.; Dobrzanski, P.; Stoehr, J.D.; Wenk, G.L. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer’s disease. Brain Res. 1998, 780, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Hauss-Wegrzyniak, B.; Vraniak, P.D.; Wenk, G.L. LPS-induced neuroinflammatory effects do not recover with time. Neuroreport 2000, 11, 1759–1763. [Google Scholar] [CrossRef] [PubMed]
- Rosi, S.; Ramirez-Amaya, V.; Hauss-Wegrzyniak, B.; Wenk, G.L. Chronic brain inflammation leads to a decline in hippocampal NMDA-R1 receptors. J. Neuroinflammation. 2004, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belarbi, K.; Jopson, T.; Tweedie, D.; Arellano, C.; Luo, W.; Greig, N.H.; Rosi, S. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J. Neuroinflamm. 2012, 9, 23–23. [Google Scholar] [CrossRef]
- Tobinick, E. Tumour necrosis factor modulation for treatment of Alzheimer’s disease: Rationale and current evidence. CNS Drugs 2009, 23, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.L.; Shi, J.X.; Hang, C.H.; Zhang, F.F.; Gao, J.; Yin, H.X. Expression of Toll-like receptor 4 in the brain in a rabbit experimental subarachnoid haemorrhage model. Inflamm. Res. 2007, 56, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.L.; Wu, W.; Ding, Y.S.; Zhang, F.F.; Hang, C.H.; Wang, H.D.; Cheng, H.L.; Yin, H.X.; Shi, J.X. Expression of Toll-like receptor 4 in the basilar artery after experimental subarachnoid hemorrhage in rabbits: A preliminary study. Brain Res. 2007, 1173, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Yin, W.N.; Cai, B.W.; Wu, J.; Wang, J.Y.; He, M.; Sun, H.; Ding, J.L.; You, C. Toll-like receptor 4/nuclear factor-κ B signaling detected in brain after early subarachnoid hemorrhage. Chin. Med. J. 2009, 122, 1575–1581. [Google Scholar] [PubMed]
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10. [Google Scholar] [CrossRef]
- Kurki, M.I.; Hakkinen, S.K.; Frosen, J.; Tulamo, R.; von und zu Fraunberg, M.; Wong, G.; Tromp, G.; Niemela, M.; Hernesniemi, J.; Jaaskelainen, J.E.; Yla-Herttuala, S. Upregulated signaling pathways in ruptured human saccular intracranial aneurysm wall: An emerging regulative role of Toll-like receptor signaling and nuclear factor-κB, hypoxia-inducible factor-1A, and ETS transcription factors. Neurosurgery 2011, 68, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tang, K.; Huang, R.Q.; Zhuang, Z.; Cheng, H.L.; Yin, H.X.; Shi, J.X. Therapeutic potential of peroxisome proliferator-activated receptor γ agonist rosiglitazone in cerebral vasospasm after a rat experimental subarachnoid hemorrhage model. J. Neurol. Sci. 2011, 305, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, S.; Laher, I. Signaling mechanisms in cerebral vasospasm. Trends Cardiovasc. Med. 2005, 15, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.H.; Cox, M.E.; Woo-Rasberry, V.; Hasty, D.L. Pathways involved in the synergistic activation of macrophages by lipoteichoic acid and hemoglobin. PLoS One 2012, 7, e47333. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Zhou, Y.; Fang, H.; Lin, S.; Wang, P.F.; Xiong, R.P.; Chen, J.; Xiong, X.Y.; Lv, F.L.; Liang, Q.L.; Yang, Q.W. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann. Neurol. 2014, 75, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.; Damore, G.; Costa, B.; Gioannini, T.L.; Weiss, J.P.; Peri, F. Hemin and a metabolic derivative coprohemin modulate the TLR4 pathway differently through different molecular targets. Innate Immun. 2011, 17, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, X.D.; Zhuang, Z.; Xue, Y.J.; Cheng, H.L.; Yin, H.X.; Shi, J.X. Peroxisome proliferator-activated receptor γ agonist rosiglitazone attenuates oxyhemoglobin-induced Toll-like receptor 4 expression in vascular smooth muscle cells. Brain Res. 2010, 1322, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Gram, M.; Sveinsdottir, S.; Ruscher, K.; Hansson, S.R.; Cinthio, M.; Akerstrom, B.; Ley, D. Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J. Neuroinflamm. 2013, 10, 1742–2094. [Google Scholar] [CrossRef]
- Spickler, E.; Lufkin, R.; Teresi, L.; Frazee, J.; Vinuela, F.; Dion, J.; Bentson, J. MR imaging of acute subarachnoid hemorrhage. Comput. Med. Imaging Graph 1990, 14, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Tsan, M.F. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor α release by murine macrophages. J. Biol. Chem. 2003, 278, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S.; Gottschalk, M.; Grenier, D. Hemoglobin and Streptococcus suis cell wall act in synergy to potentiate the inflammatory response of monocyte-derived macrophages. Innate Immun. 2008, 14, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Elmer, J.; Harris, D.; Palmer, A.F. Purification of hemoglobin from red blood cells using tangential flow filtration and immobilized metal ion affinity chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 131–138. [Google Scholar] [CrossRef]
- Tsan, M.F.; Gao, B. Endogenous ligands of Toll-like receptors. J. Leukoc. Biol. 2004, 76, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Tsan, M.F. Heat shock proteins and high mobility group box 1 protein lack cytokine function. J. Leukoc. Biol. 2011, 89, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, S. Toll-like receptors and inflammation in the CNS. Curr. Drug Targets Inflamm. Allergy 2002, 1, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.L.; O’Neill, L.A. Toll-like receptors: From the discovery of NFκB to new insights into transcriptional regulations in innate immunity. Biochem. Pharmacol. 2006, 72, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Rossol, M.; Heine, H.; Meusch, U.; Quandt, D.; Klein, C.; Sweet, M.J.; Hauschildt, S. LPS-induced cytokine production in human monocytes and macrophages. Crit. Rev. Immunol. 2011, 31, 379–446. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Li, G.; Qian, X.; Liu, Y.; Wu, X.; Liu, B.; Hong, J.S.; Block, M.L. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia 2005, 52, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lapointe, B.M.; Clark, S.R.; Zbytnuik, L.; Kubes, P. A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J. Immunol. 2006, 177, 8103–8110. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Li, M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Aida, Y.; Kusumoto, K.; Nakatomi, K.; Takada, H.; Pabst, M.J.; Maeda, K. An analogue of lipid A and LPS from Rhodobacter sphaeroides inhibits neutrophil responses to LPS by blocking receptor recognition of LPS and by depleting LPS-binding protein in plasma. J. Leukoc. Biol. 1995, 58, 675–682. [Google Scholar] [PubMed]
- Lu, Z.; Zhang, X.; Li, Y.; Jin, J.; Huang, Y. TLR4 antagonist reduces early-stage atherosclerosis in diabetic apolipoprotein E-deficient mice. J. Endocrinol. 2013, 216, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Jerala, R. Structural biology of the LPS recognition. Int. J. Med. Microbiol. 2007, 297, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, J.Y. Anti-CD14 antibody reduces LPS responsiveness via TLR4 internalization in human monocytes. Mol. Immunol. 2014, 57, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef] [PubMed]
- Leturcq, D.J.; Moriarty, A.M.; Talbott, G.; Winn, R.K.; Martin, T.R.; Ulevitch, R.J. Antibodies against CD14 protect primates from endotoxin-induced shock. J. Clin. Investig. 1996, 98, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Schimke, J.; Mathison, J.; Morgiewicz, J.; Ulevitch, R.J. Anti-CD14 mAb treatment provides therapeutic benefit after in vivo exposure to endotoxin. Proc. Natl. Acad. Sci. USA 1998, 95, 13875–13880. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Ogasawara, T.; Homma, T.; Saito, H.; Matsumoto, K. Lipopolysaccharide-binding protein critically regulates lipopolysaccharide-induced IFN-β signaling pathway in human monocytes. J. Immunol. 2004, 172, 6185–6194. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Tosun, C.; Ivanova, S.; Kurland, D.B.; Hong, C.; Radecki, L.; Gisriel, C.; Mehta, R.; Schreibman, D.; Gerzanich, V. Heparin reduces neuroinflammation and transsynaptic neuronal apoptosis in a model of subarachnoid hemorrhage. Transl. Stroke Res. 2012, 3, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Tosun, C.; Kurland, D.B.; Mehta, R.; Castellani, R.J.; deJong, J.L.; Kwon, M.S.; Woo, S.K.; Gerzanich, V.; Simard, J.M. Inhibition of the Sur1-Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke 2013, 44, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Zoerle, T.; Ilodigwe, D.; Wan, H.; Lakovic, K.; Sabri, M.; Ai, J.; Macdonald, R.L. Pharmacologic reduction of angiographic vasospasm in experimental subarachnoid hemorrhage: Systematic review. Acta Neurochir. Suppl. 2013, 115, 247–251. [Google Scholar] [PubMed]
- Fassbender, K.; Hodapp, B.; Rossol, S.; Bertsch, T.; Schmeck, J.; Schutt, S.; Fritzinger, M.; Horn, P.; Vajkoczy, P.; Wendel-Wellner, M.; et al. Endothelin-1 in subarachnoid hemorrhage: An acute-phase reactant produced by cerebrospinal fluid leukocytes. Stroke 2000, 31, 2971–2975. [Google Scholar] [CrossRef] [PubMed]
- Sehba, F.A.; Bederson, J.B. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol. Res. 2006, 28, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Sehba, F.A.; Pluta, R.M.; Zhang, J.H. Metamorphosis of subarachnoid hemorrhage research: From delayed vasospasm to early brain injury. Mol. Neurobiol. 2011, 43, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Yan, J.; Rolland, W.B.; Soejima, Y.; Caner, B.; Zhang, J.H. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl. Stroke Res. 2013, 4, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Sabri, M.; Lass, E.; Macdonald, R.L. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res. Treat. 2013, 2013. [Google Scholar] [CrossRef]
- Simard, J.M.; Schreibman, D.; Aldrich, E.F.; Stallmeyer, B.; Le, B.; James, R.F.; Beaty, N. Unfractionated heparin: Multitargeted therapy for delayed neurological deficits induced by subarachnoid hemorrhage. Neurocrit. Care 2010, 13, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sagher, O.; Keep, R.; Hua, Y.; Xi, G. Comparison of experimental rat models of early brain injury after subarachnoid hemorrhage. Neurosurgery 2009, 65, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Weir, B.K. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke 1991, 22, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Lisk, C.; Kominsky, D.; Ehrentraut, S.; Bonaventura, J.; Nuss, R.; Hassell, K.; Nozik-Grayck, E.; Irwin, D.C. Hemoglobin-induced endothelial cell permeability is controlled, in part, via a myeloid differentiation primary response gene-88-dependent signaling mechanism. Am. J. Respir. Cell. Mol. Biol. 2013, 49, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.; Jeney, V.; Chora, A.; Larsen, R.; Balla, J.; Soares, M.P. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J. Biol. Chem. 2009, 284, 29582–29595. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Keep, R.F.; He, Y.; Sagher, O.; Hua, Y.; Xi, G. Hemoglobin and iron handling in brain after subarachnoid hemorrhage and the effect of deferoxamine on early brain injury. J. Cereb. Blood Flow Metab. 2010, 30, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.F.; Sun, G.; Harris, D.R. Tangential flow filtration of hemoglobin. Biotechnol. Prog. 2009, 25, 189–199. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin Is an Endogenous Toll-Like Receptor 4 Ligand—Relevance to Subarachnoid Hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028-5046. https://doi.org/10.3390/ijms16035028
Kwon MS, Woo SK, Kurland DB, Yoon SH, Palmer AF, Banerjee U, Iqbal S, Ivanova S, Gerzanich V, Simard JM. Methemoglobin Is an Endogenous Toll-Like Receptor 4 Ligand—Relevance to Subarachnoid Hemorrhage. International Journal of Molecular Sciences. 2015; 16(3):5028-5046. https://doi.org/10.3390/ijms16035028
Chicago/Turabian StyleKwon, Min Seong, Seung Kyoon Woo, David B. Kurland, Sung Hwan Yoon, Andre F. Palmer, Uddyalok Banerjee, Sana Iqbal, Svetlana Ivanova, Volodymyr Gerzanich, and J. Marc Simard. 2015. "Methemoglobin Is an Endogenous Toll-Like Receptor 4 Ligand—Relevance to Subarachnoid Hemorrhage" International Journal of Molecular Sciences 16, no. 3: 5028-5046. https://doi.org/10.3390/ijms16035028