1. Introduction
Myocardial infarction (MI) remains a major cause of morbidity and mortality despite that marked improvements in medical and device therapy have been achieved [
1]. MI causes the dysfunction of cardiomyocytes, enhancement of vascular permeability, myocardial fibrosis and left ventricular remodeling. Indeed, cardiomyocyte apoptosis and vascular permeability are involved in myocardial fibrosis and LV remodeling, thus promoting contractile dysfunction [
2]. After these effects, irreversible damage occurs in the ischemic heart.
Pigment epithelium-derived factor (PEDF), a non-inhibitory member of the serine protease inhibitor superfamily (SERPINS), is a glycoprotein with a molecular weight of 50 kDa identified in various human tissues, such as eye, brain, muscle, adipose tissue, liver and bone [
3,
4,
5]. PEDF is characterized as a multifunctional protein possessing antiangiogenic, antitumorigenic, antioxidant, anti-inflammatory, antithrombotic, neurotrophic and neuroprotective properties [
6,
7,
8,
9]. PEDF regulates vascular permeability by preventing the dissociation of endothelial adherens junctions (AJs) and tight junctions (TJs) via a γ-secretase-dependent mechanism in retinal microvascular endothelial cells [
10]. Recently, PEDF mRNA and protein have been demonstrated to be expressed in the human heart and secreted by cardiomyocytes and fibroblasts [
11]. PEDF may rely on any of its antioxidant, anti-inflammatory and antithrombotic protective properties to manifest a counteractive mechanism during the development of atherosclerosis and cardiovascular disease [
12].
The findings mentioned above led us to speculate that PEDF could exert beneficial effects on cardiac function by suppressing vascular permeability and cardiomyocyte apoptosis in acute myocardial infarction (AMI). However, the protective role of PEDF against vascular permeability after AMI remains unknown, and some investigations about the effect of PEDF on cardiomyocytes were controversial. Ueda, S.
et al. [
13] demonstrated that PEDF inhibits tissue remodeling and improves cardiac function in the rat AMI model by reducing the death of apoptotic cells, oxidative stress generation and suppressing cardiac fibrosis. Nevertheless, Li
et al. [
14] found that PEDF increased cardiomyocyte apoptosis during hypoxia via the Fas signaling pathway. Kathrin Rychli’s study revealed that PEDF may contribute to the progression of heart failure by inducing apoptosis in human cardiomyocytes and fibroblasts via activation of caspase-3 [
15].
Therefore, in this study, we delivered lentivirus carrying PEDF or PEDF RNAi by using intramyocardial injections to overexpress or knockdown PEDF in a rat AMI model. We aimed to investigate whether PEDF could inhibit vascular permeability and cardiomyocyte apoptosis and improve cardiac function in rats with AMI and tried to elucidate the possible mechanisms.
3. Discussion
Recent investigations have suggested that PEDF treatment can potently inhibit tissue remodeling and improve cardiac function in the rat AMI model [
13] and suppress retinal microvascular permeability [
10]. However, to date, it is not known whether and how PEDF acts to inhibit hypoxic or ischemic endothelial injury in the heart. Meanwhile, some investigations about the effect of PEDF on cardiomyocytes were controversial. In this study, we reported that although PEDF inhibited angiogenesis in the infarct border zone, PEDF reduced vascular permeability and infarct size, protected cardiomyocytes in an ischemic heart and improved functional recovery in an AMI rat model. We also discovered a new and beneficial role for PEDF, which prevented hypoxia and ischemia-induced endothelial barrier dysfunction and revealed the molecular basis for PEDF-mediated protection of hypoxic RAECs. To the best of our knowledge, this is the first report delineating the role of PEDF in the regulation of vascular permeability in RAECs via PPARγ under the hypoxic condition. In addition, we found that PEDF protected cardiomyocytes against ischemia or hypoxia-induced cell apoptosis, both
in vivo and
in vitro, via preventing the activation of caspase-3.
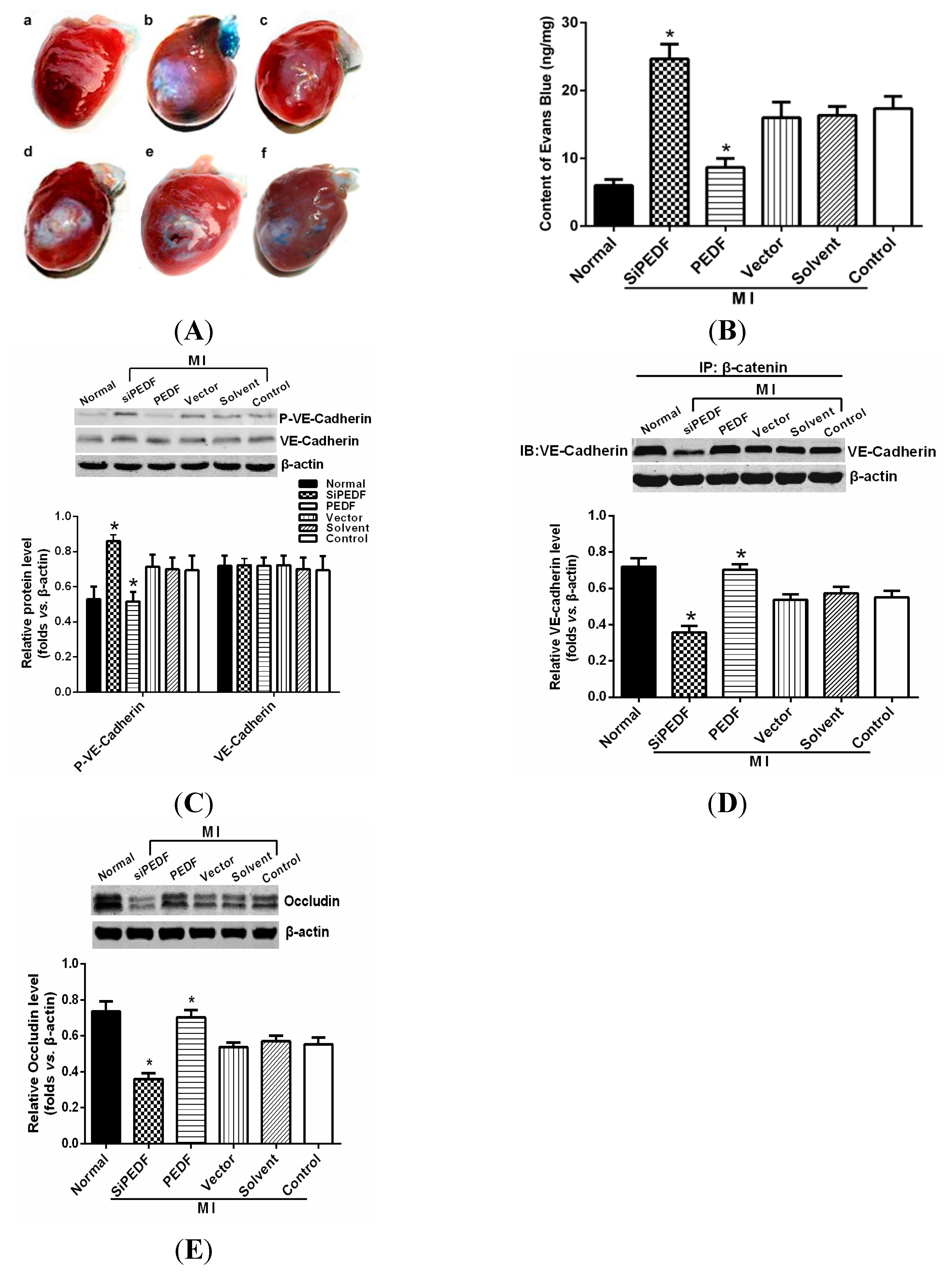
We observed that PEDF was downregulated in the peri-infarct region after AMI. Thus, to verify the role of PEDF in ischemic heart, we delivered lentivirus carrying PEDF or PEDF RNAi by using intramyocardial injections to overexpress or knockdown PEDF in a rat AMI model (
Supplementary Figure S1). In ischemic heart, the vascular permeability was increased with vascular perfusion dysfunction, which was not conducive to functional recovery of AMI. We found that silencing of PEDF significantly decreased the binding of VE-cadherin with β-catenin and the expression of occludin, resulting in destabilized endothelial junctions and high vascular permeability in an AMI rat model. Our results are consistent with the studies that PEDF inhibits vascular permeability in retinal micro-vessels [
10]. Thus, although PEDF had a negative effect on angiogenesis, it effectively reduced local vascular permeability, indicating that PEDF may improve functional recovery in the ischemic heart.
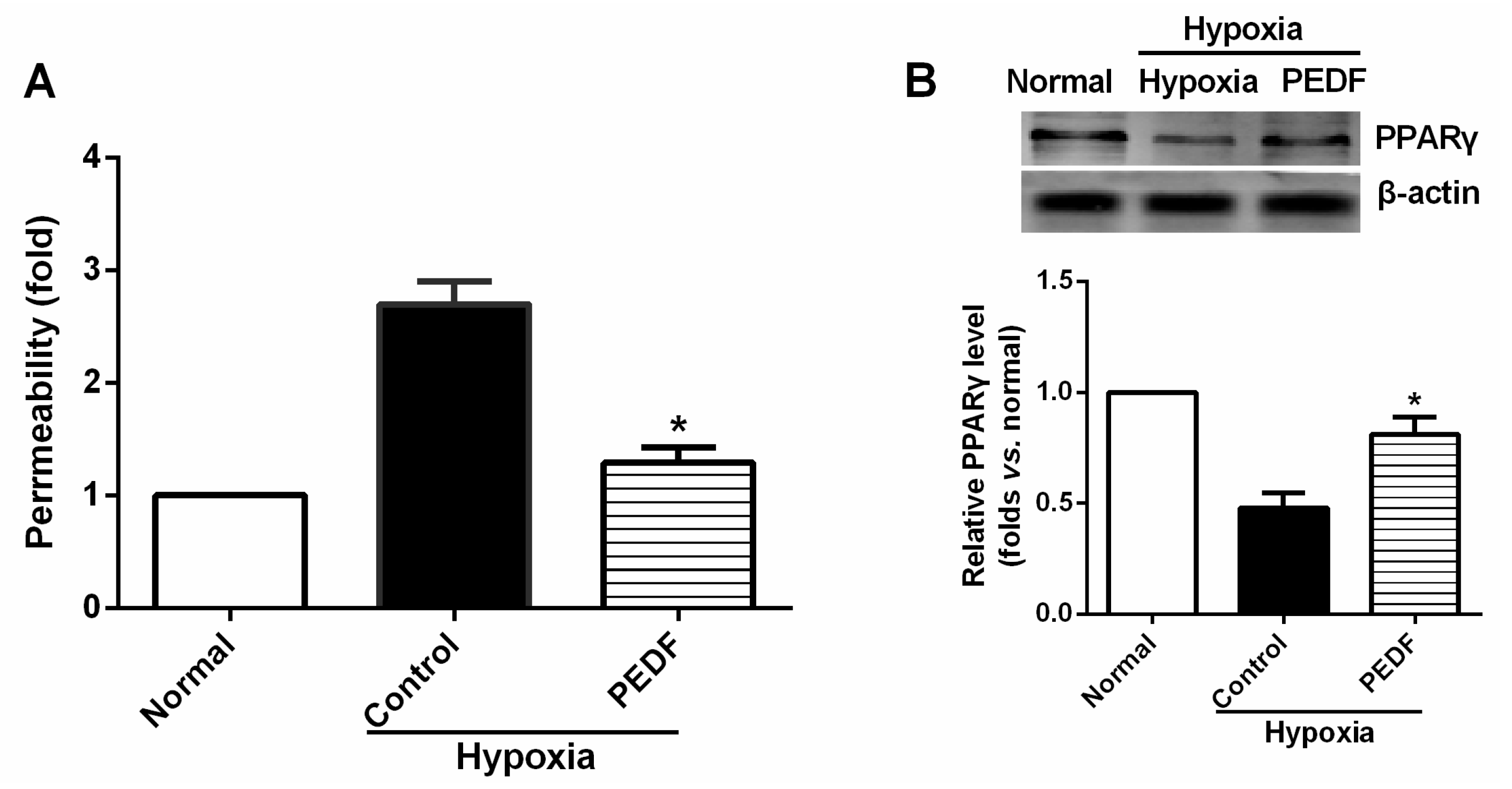
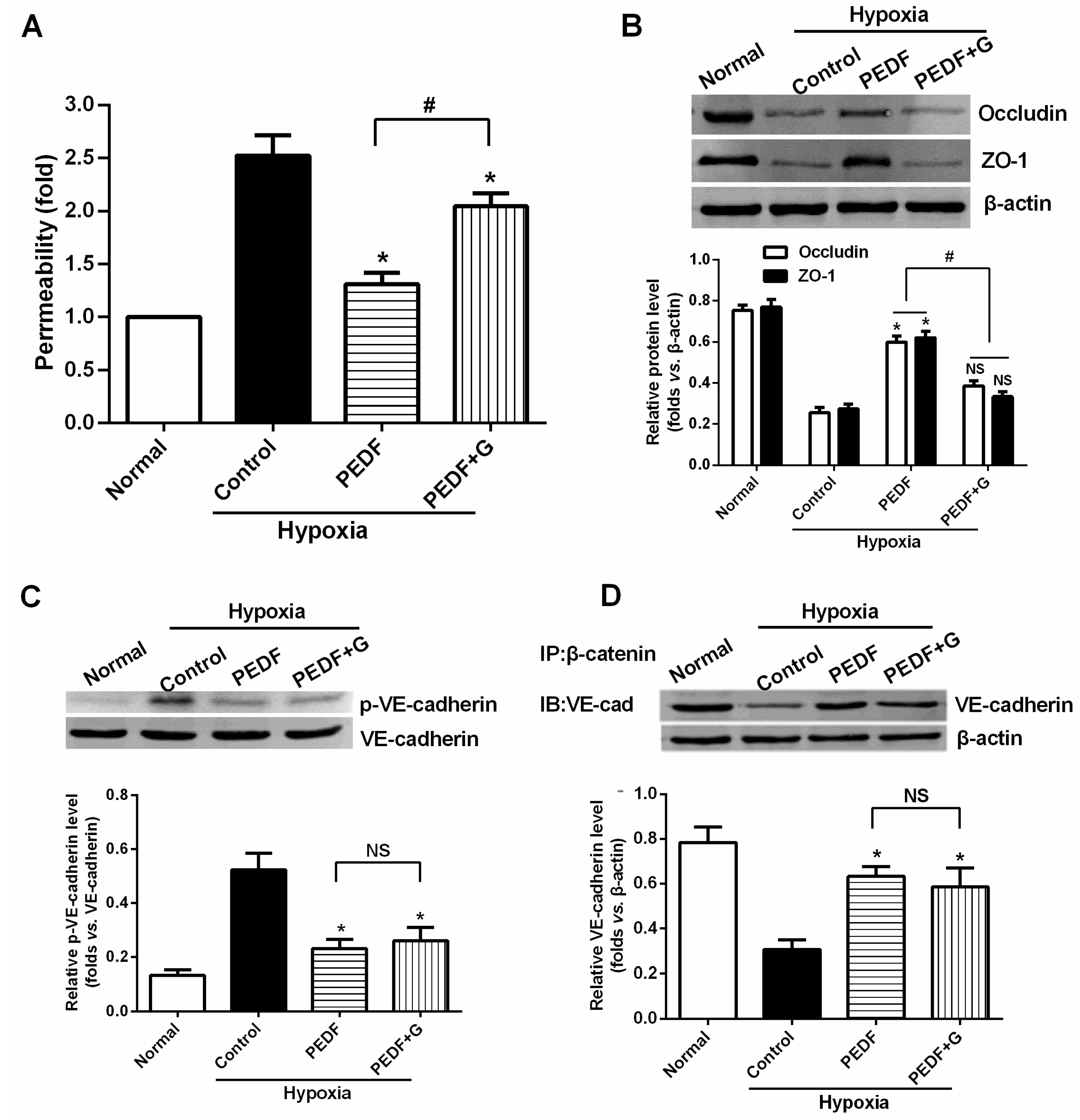
The most important finding of this study is that PEDF-mediated PPARγ production in hypoxic RAECs promotes the upregulation of TJs, leading to a sustained barrier function. Such observations may be important to ensure that PPARγ is an important regulator of the signaling pathway by which PEDF inhibits endothelial permeability. Although the exact mechanism remains elusive, some recent studies have suggested that activation of PPARγ stimulates lipid synthesis, epidermal differentiation and aquaporin 3 expression and accelerates recovery from permeability barrier dysfunction [
19]. PPARγ agonists upregulate the barrier function of tight junctions via a PKC pathway in human nasal epithelial cells [
17]. Furthermore, when we added PPARγ inhibitor GW9662, PEDF was unable to suppress hypoxia-promoted permeability effectively in RAECs, and PEDF-mediated upregulation of occludin and ZO-1 proteins retracted. Interestingly, PEDF’s effects on the AJs were nearly unchanged in the presence of PPARγ inhibitor GW9662. These findings suggest that PEDF inhibits RAEC permeability under hypoxia via enhancing the expression of TJs and AJs. However, TJs may represent a major role by which PEDF inhibits RAEC permeability through PPARγ. The effects of PEDF upregulating AJs may be via the other path. Consequently, further study is needed to address the issue.
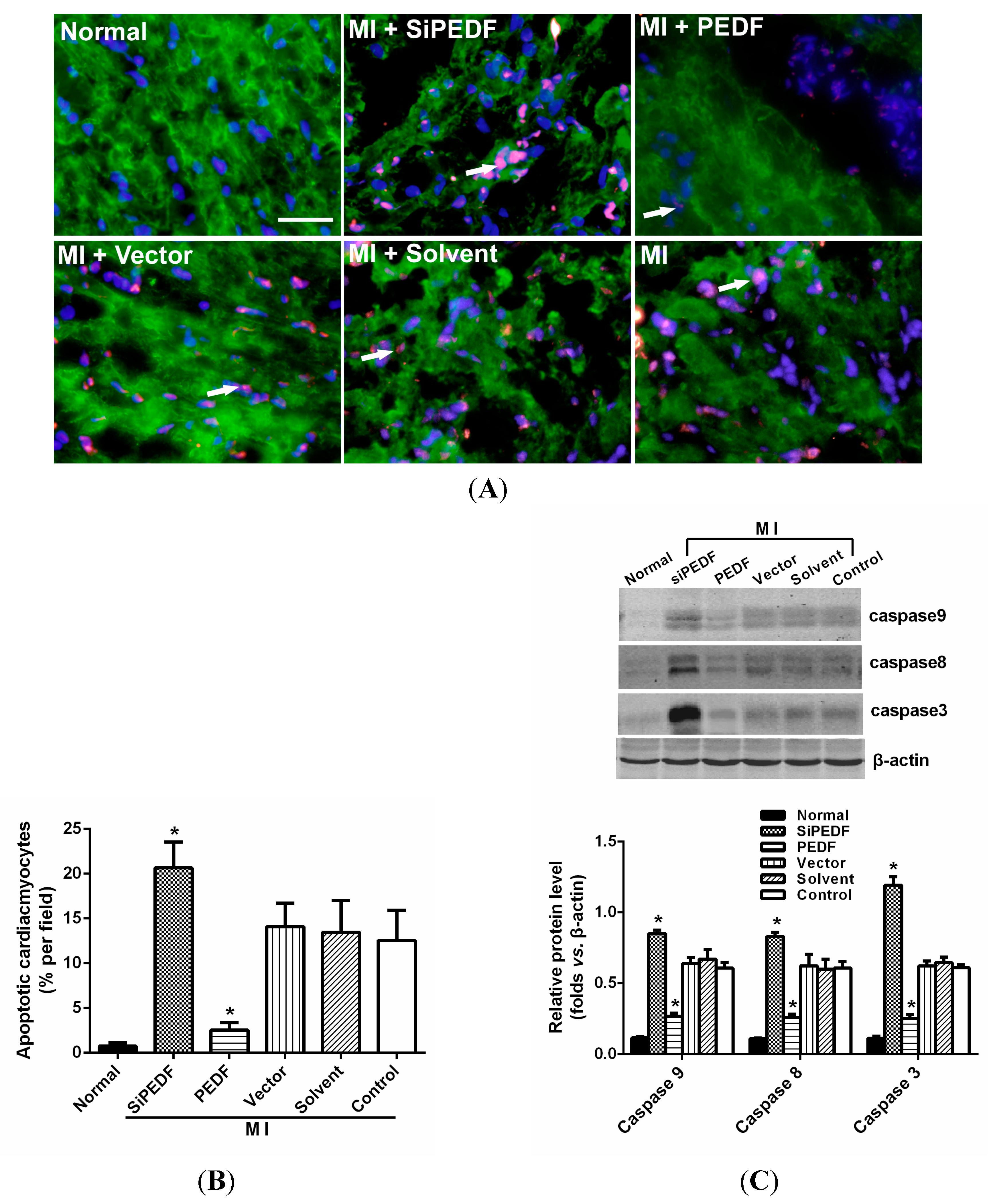
It is widely recognized that cardiomyocyte death is an important component in the pathogenesis of myocardial infarction and heart failure [
20]. Pathologically, failing hearts after AMI exhibit ongoing cardiomyocyte death over months to years at levels that are low, but still 100-fold higher than those observed in normal hearts [
21]. Our study showed that knocking down PEDF increased cardiomyocyte apoptosis, whereas PEDF overexpression decreased the apoptosis. The expression levels of FASL and caspase-8, crucial triggers in the death receptor domain signal pathway, were increased in the peri-infarct region after initiating AMI. Meanwhile, the expression of caspase-9 was also increased, which subsequently activated the mitochondrial apoptotic pathway. We found that the activation of these two apoptotic pathways was remarkably enhanced once PEDF expression was silenced. In contrast, PEDF overexpression effectively inhibited these two apoptotic pathways, thus presenting an anti-apoptotic property. Our results demonstrate that PEDF may enhance cardiomyocyte survival by protecting cardiomyocytes against apoptosis in the ischemic heart.
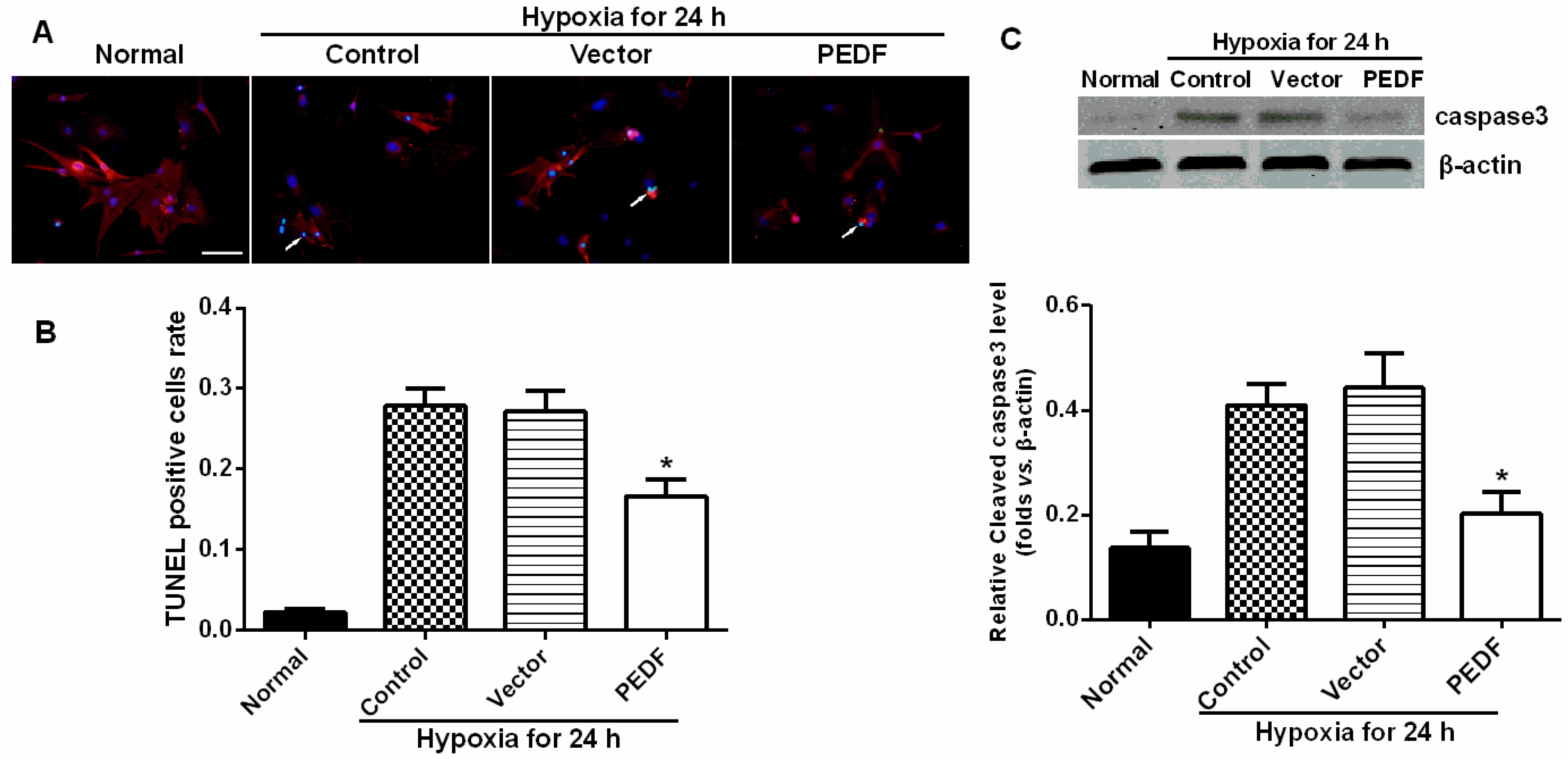
In addition, we also found that PEDF protected cardiomyocytes against hypoxia-induced cell apoptosis via preventing the activation of caspase-3. However, Li
et al. [
14] reported that PEDF (200 ng/mL) increased cardiomyocyte apoptosis under both hypoxic and normoxic conditions. This conclusion conflicts with Ueda’s finding [
13]
, and Ueda
et al. demonstrated that a concentration of PEDF close to 200 ng/mL in normal serum caused no impairment of cardiomyocyte survival. Recently, several studies presented proofs of concept of yet another therapeutic role for PEDF, that of retinal neuroprotection from oxidative stress, and provided evidence of a potential effect on the broader area of apoptosis-mediated cell death [
22]. Oxidative stress is also a mediator of cardiomyocyte apoptosis. Our previous studies also found that PEDF protected cultured H9c2 cells against apoptosis and necroptosis under hypoxic condition via the anti-oxidative mechanism [
23]. The result matches that of the
in vitro experiments in this study.
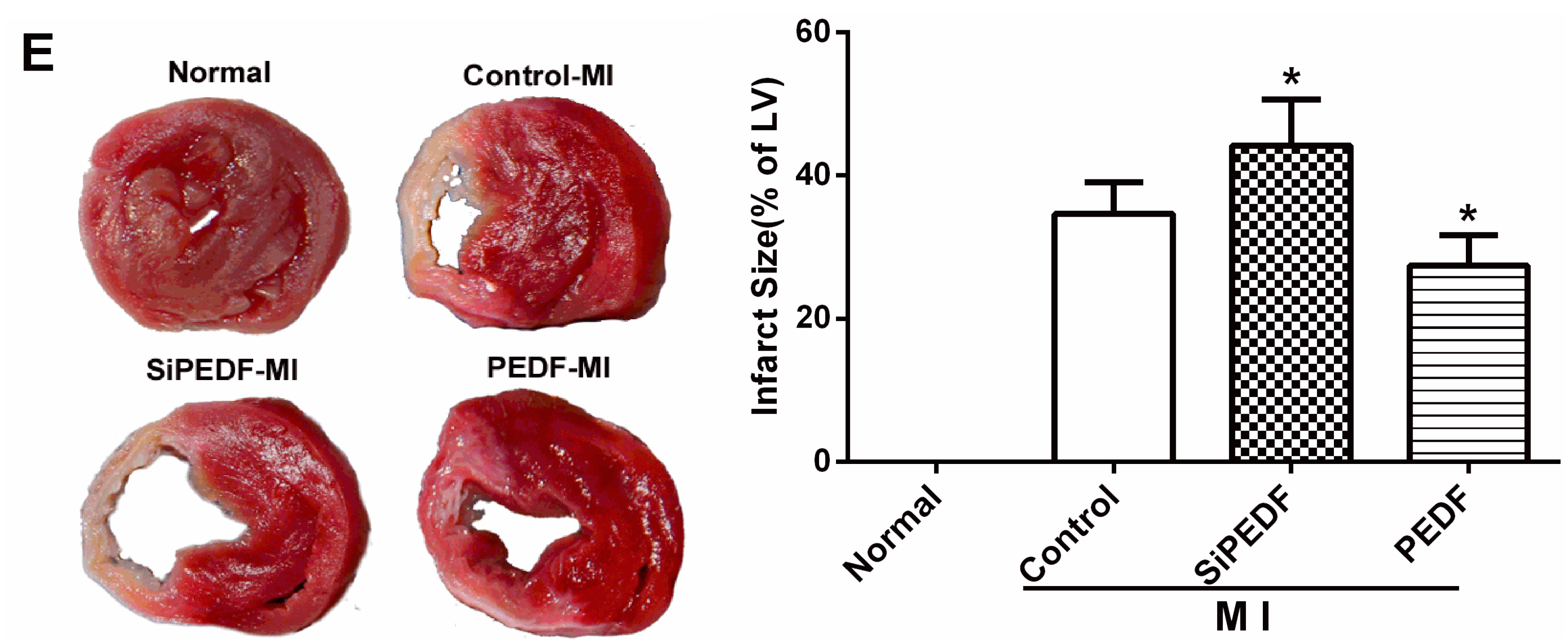
There may be some relationship between the events of vascular permeability and myocardial apoptosis. The effect of anti-permeability to inhibit cardiac inflammation and maintain the cardiac environment may represent an important mechanism that further links cardiomyocyte protection to cardiovascular disease. Furthermore, we evidenced that PEDF reduced the infarct size, resulting in functional recovery in an AMI rat model. Given the fact that PEDF levels were decreased in the peri-infarct zone, our present study provides a novel therapeutic potential of the substitution of PEDF for cardiac dysfunction in AMI.
4. Experimental Section
4.1. Recombinant Lentivirus Constructs and Viral Production
Recombinant lentivirus (LV) was prepared as described previously. PEDF over-expression plasmids and the RNAi vector PEDF-RNAi-LV of the PEDF gene producing PEDF shRNA were successfully constructed and were then successfully packaged in 293T cells. The concentrated titer of virus suspension was 2 × 1012 TU/L.
4.2. Animal Model and Intramyocardial Gene Delivery
Sprague-Dawley male rats (8 to 10 weeks old, weighing 210–250 g, 230 ± 20 g) were obtained from the Experimental Animal Center of Xuzhou Medical College, Xuzhou, China. All of the experiments conform to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication, 8th Edition, 2011). The animal care and experimental protocols were approved by the Xuzhou Medical College Committee on Animal Care.
Myocardial ischemia was induced by ligation of the left-anterior descending coronary artery (LAD) in anesthetized rats, as described previously [
24] (see the
Supplementary Material, Methods 1.1). For intramyocardial gene delivery, PEDF-LV or PEDF-RNAi-LV (2 × 10
7 TU) in 20 μL enhanced infection solution (ENIS, pH 7.4) was delivered with a 20-μL syringe and 25-gauge needle into the myocardium along the infarct border immediately after surgery. Control animals received an equivalent volume of either ENIS or lentivirus vector. The reliability of the injection transfection method was confirmed by the blue-stained myocardial size infiltrated by injected methylene blue (
Supplementary Figure S2). The animal models were randomly divided into six groups: Group A (normal); Group B (siPEDF), PEDF-RNAi-LV was transferred after surgery; Group C (PEDF), PEDF-LV was transferred after surgery; Group D (vector control), LV vector was transferred into the ischemic myocardium; Group E (solvent control), 20 μL ENIS were transferred as a solvent control; and Group F (ischemic control), the animal did not undergo any gene transfer after surgery. The animals were sacrificed with an overdose of sodium pentobarbitone (60 mg/kg, i.v.), and their hearts were harvested at the end of 2 or 4 weeks after AMI for further analysis.
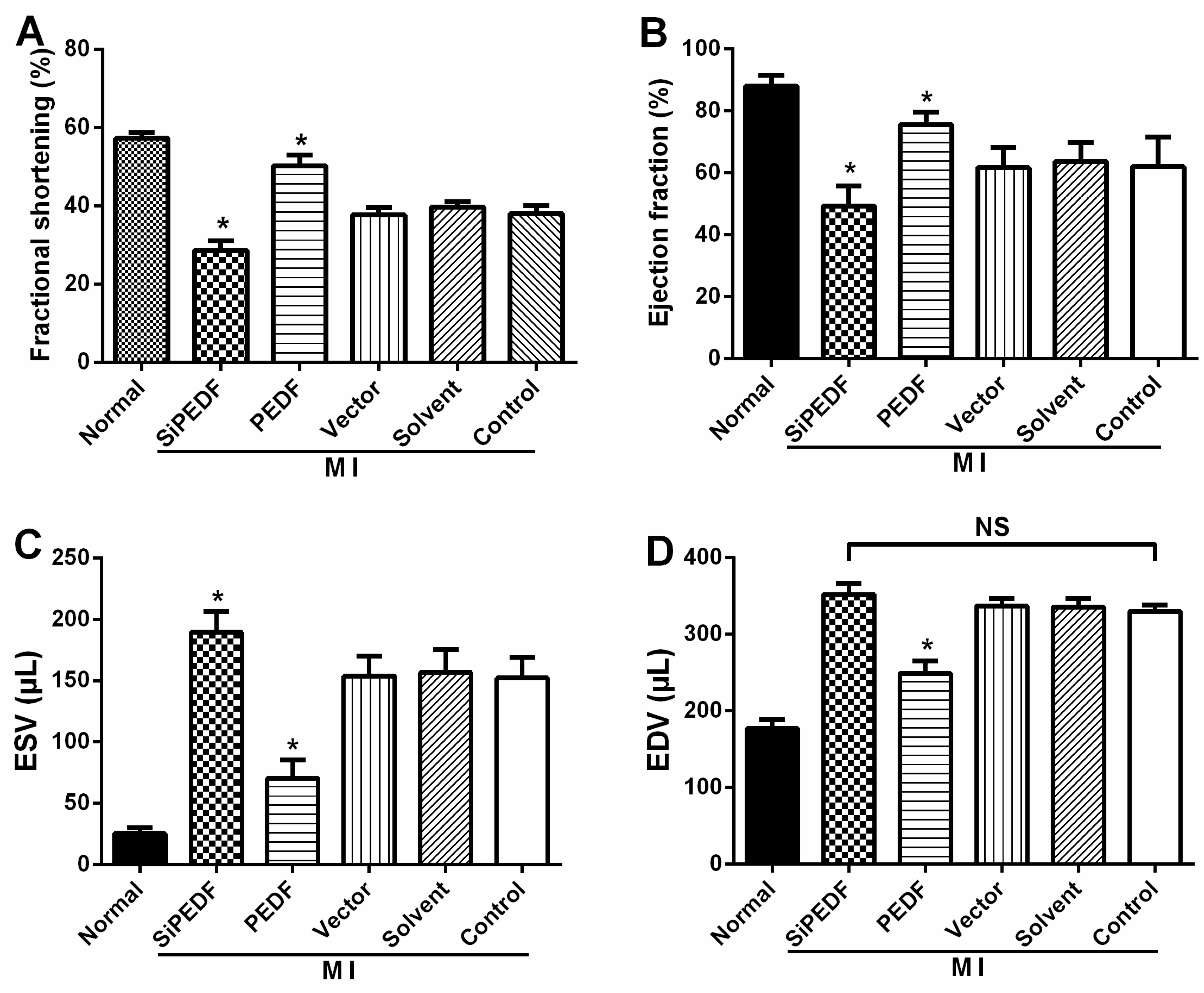
4.3. Animal Cardiac Function Evaluation
Echocardiography was conducted under sedation by sodium pentobarbital (30 mg/kg, i.p), as described previously [
25]. Two-dimensional-guided (2D) M-mode echocardiography was used to determine left ventricular (LV) chamber volume at systole and diastole and contractile parameters, such as left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic dimension (LVESD), left ventricular end-diastolic volume (LVEDV) and left ventricular end-systolic volume (LVESV). The left ventricular fractional shortening (LVFS) was calculated as follows: FS (%) = (LVEDD − LVESD)/LVEDD × 100. The ejection fraction (EF) was then derived as EF (%) = (EDV – ESV)/EDV × 100.
4.4. Quantification of Myocardial Infarct Size
Four weeks after LAD artery ligation, the selected rats were euthanized, then the hearts were removed for MIS analyses by the method of 2,3,5-triphenyltetrazolium (TTC) staining [
26]. The left ventricle (LV) was isolated and cut into 4-mm slices perpendicular to the axis of the LAD. Then, the slices were immediately immersed in 1% TTC (Sigma Chemical Co., St. Louis, MO, USA) in phosphate buffer (pH 7.4) at 37 °C for 10 min to discriminate the infarcted tissue from viable myocardium. All slices were scanned from both sides by a color CCD camera (FV-10, Fuji, Japan), and in each slide, the infarct area was compared with the total area by using digital planimetry software (Image-Pro Plus 6.0, Media Cybernetics Inc., Bethesda, MD, USA). After correction with the weight of the slices, the infarct size was calculated as a percentage of the LV.
4.5. Cardiomyocyte Apoptosis Assay in Vivo
The myocardial samples from the animal left ventricle were embedded in optimum cutting temperature (OCT) compound tissue medium (SAKURA, Tokyo, Japan), snap-frozen on dry ice and stored at −80 °C. Cardiomyocyte apoptosis in vivo was determined by double-labeling immunofluorescence staining of TUNEL, which was performed with an in situ cell death detection kit according to the manufacturer’s instructions using the In Situ Cell Death Detection Kit, POD (Cat. No, 11684817910, Roche Applied Science, Mannheim, Germany). Cardiomyocytes were identified using α-sarcomeric actinin antibodies (α-sa) (Sigma Chemicals). Hoechst (Sigma Aldrich) staining was used to count the total number of nuclei. Cardiomyocyte nuclei with a larger diameter can be recognized as being located within myofibers. Cardiomyocytes from at least three randomly selected sections or 48-well plates were evaluated for these variables. The number of TUNEL-positive cells was calculated as cells per area at 400-fold magnification. The percentage of TUNEL-positive cells was calculated as a percentage of total cells viewed in five randomly selected fields for each group.
4.6. Vascular Permeability Measurement
To determine vascular permeability in the rat hearts, a Miles assay was employed based on the method described previously [
27,
28]. Two percent Evans Blue dye (30 mg/kg, Sigma Chemicals) was injected intravenously 30 min before animal sacrifice. After sacrifice, the sample of heart tissue was weighed, and the Evans Blue dye was extracted by immersion in formamide (1 mL/100 mg) overnight at 60 °C. The extract was centrifuged at a speed of 7000 rpm for 45 min at 4 °C. The content of Evans Blue dye was determined on the basis of absorbance at 620 nm, and the ratio between the transferred and the intact normal heart samples was calculated and normalized to the tissue weight.
4.7. Western Blot Analysis and Co-Immunoprecipitation
Total proteins were extracted from the myocardium of the peri-infarct zone with ice-cold lysis buffer, as described previously [
27]. Membranes were incubated with the following primary antibodies: PEDF, TNFα, IL-6 and FasL (Santa Cruz Biotechnology, Santa Cruz, CA, USA), ZO-1, p-VE-cadherin (Abcam, Cambridge, MA, USA); β-catenin, VE-cadherin, occludin, caspase-9, caspase-8 and caspase-3 (Anbo Biotechnology Co., Ltd., San Francisco, CA, USA). β-actin was used as a loading control. Co-immunoprecipitation (co-IP) was performed to examine the combination of VE-cadherin/β-catenin, as detailed in the
Supplementary Materials (Methods 1.2).
4.8. Preparations of PEDF Protein
Recombinant rat PEDF (GenBankTM Accession Number NM_177927) was synthesized by Cusabio Biotech, Co., Ltd. (Wuhan, China). In brief, 293T cells were transfected with the recombinant vector pGEX 6P-1 GST-tagged PEDF. GST-tagged PEDF proteins were purified by high pressure liquid chromatography purification (>90% purity) and amino-terminal sequence determination. The resulting proteins were soluble in aqueous solutions.
4.9. Cardiomyocyte Culture and Cardiomyocyte Apoptosis Assay in Vitro
Neonatal Sprague-Dawley (SD) rats (1–3 days old, weighing 5–7 g, means 6.1 ± 0.7 g) were obtained from the Experimental Animal Centre of Xuzhou Medical College. All of the experiments conform to the guidelines as described before on the ethical use of animals. Ventricular myocytes were isolated from neonatal SD rats. The apoptosis assay was used to study four groups: a normal control group, a hypoxia group, a vector control (10 nmol/L) + hypoxia group and a PEDF-treated (10 nmol/L) + hypoxia group. The cardiomyocytes in the normal control group were maintained under normoxic conditions (95% air, 5% CO2) at 37 °C for 24 h. Cardiomyocytes in the hypoxia groups were exposed to hypoxic conditions (94% N2, 5% CO2, 1% O2) in an anaerobic system (Thermo Forma, Marietta, OH, USA) at 37 °C for equivalent periods. The cells were then harvested, and apoptotic cells were detected by TUNEL staining according to the manufacturer’s protocol.
4.10. RAECs Culture and Permeability Measurements in Vitro
RAECs were isolated and cultured as described previously [
29]. Changes in the permeability of monolayer RAECs were examined using cell culture transwell inserts (12 mm diameter, 0.4 mm pore size; Transwell, Corning, Cambridge, MA, USA). Endothelial cells were maintained at confluence on porous polyester membrane inserts prior to experimentation. The upper and lower compartments contained 150 and 0.5 mL of media, respectively. PEDF (10 nmol/L) was added to both the upper and the lower chambers for 1 hour before the addition of 10 μM FITC-dextran to the upper chamber of the inserts. Then, RAECs were exposed to hypoxic conditions (94% N
2, 5% CO
2, 1% O
2) in an anaerobic system (Thermo Forma) at 37 °C for 24 h, while the controls were left in normoxic conditions at 37 °C for the equivalent periods. A 50-μL sample of medium was taken in triplicate from the lower chamber and placed in 96-well cluster plates (black with clear bottoms, polystyrene; Corning Costar) to measure fluorescence intensity (excitation at 530 nm and emission at 590 nm). The FITC-dextran that passed across the endothelial cell monolayer was normalized to the fluorescence reading from the upper chamber, and permeability was calculated as relative fluorescence units.
4.11. Statistical Analyses
All of the data were expressed as the means ± SD and analyzed by analysis of variance and a Student-Newman-Keuls test using the SPSS13.0 software (SPSS Inc., Chicago, IL, USA). p-Values of less than 0.05 were considered to be statistically significant.
5. Conclusions
In this study, we discovered a new and beneficial role for PEDF, which prevented hypoxia and ischemia-induced endothelial barrier dysfunction and revealed the molecular basis for PEDF-mediated protection of hypoxic RAECs. We first reported that PEDF inhibits vascular permeability in RAECs via PPARγ under the hypoxic condition and found that PEDF protected cardiomyocytes against ischemia or hypoxia-induced cell apoptosis, both in vivo and in vitro via preventing the activation of caspase-3. PEDF may rely on any of its anti-leak and anti-apoptotic protective properties to reduce myocardial infarct size and to protect cardiac function during the development of ischemic injury in an AMI rat model. PEDF may be an indispensable environmental factor in stabilizing the cardiac microenvironment and enhancing performance of the ischemic heart. Our findings may provide a novel therapeutic strategy for ischemic heart disease.
A limitation of the study is that these results only indicated the biological effects of PEDF on cardiomyocyte apoptosis and endothelial cell permeability, the connection between endothelial cell permeability and cardiomyocyte apoptosis and what the role of PEDF in this connection is are uncertain. In addition, the effects of PEDF upregulating AJs need further study; the detailed molecular mechanisms are not certain here. Therefore, future investigation is needed to clarify these detailed mechanisms underlying the cardiac protection initiated by PEDF in the ischemic heart.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






