
SOD2 Activity Is not Impacted by Hyperoxia in Murine Neonatal Pulmonary Artery Smooth Muscle Cells and Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
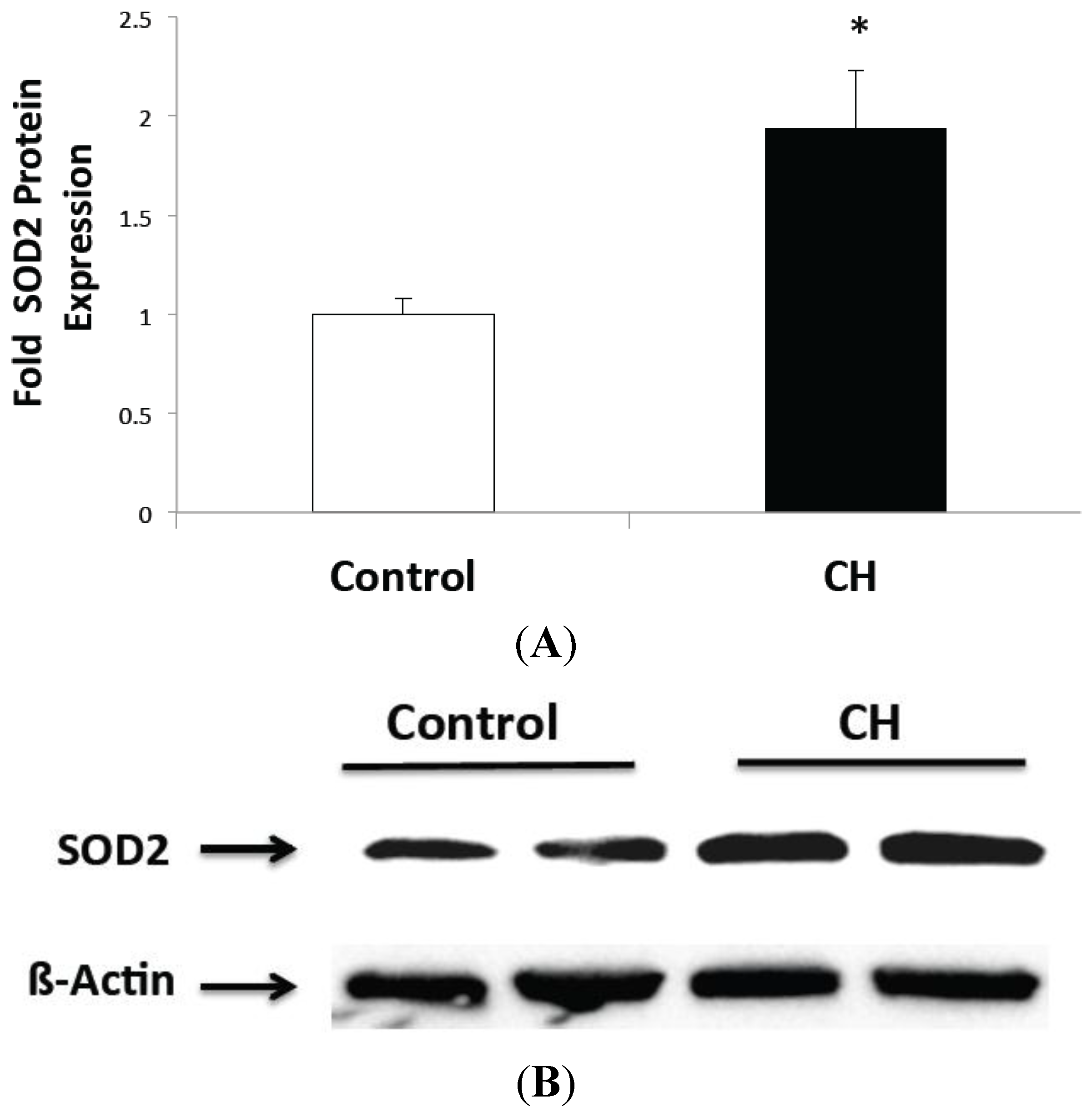
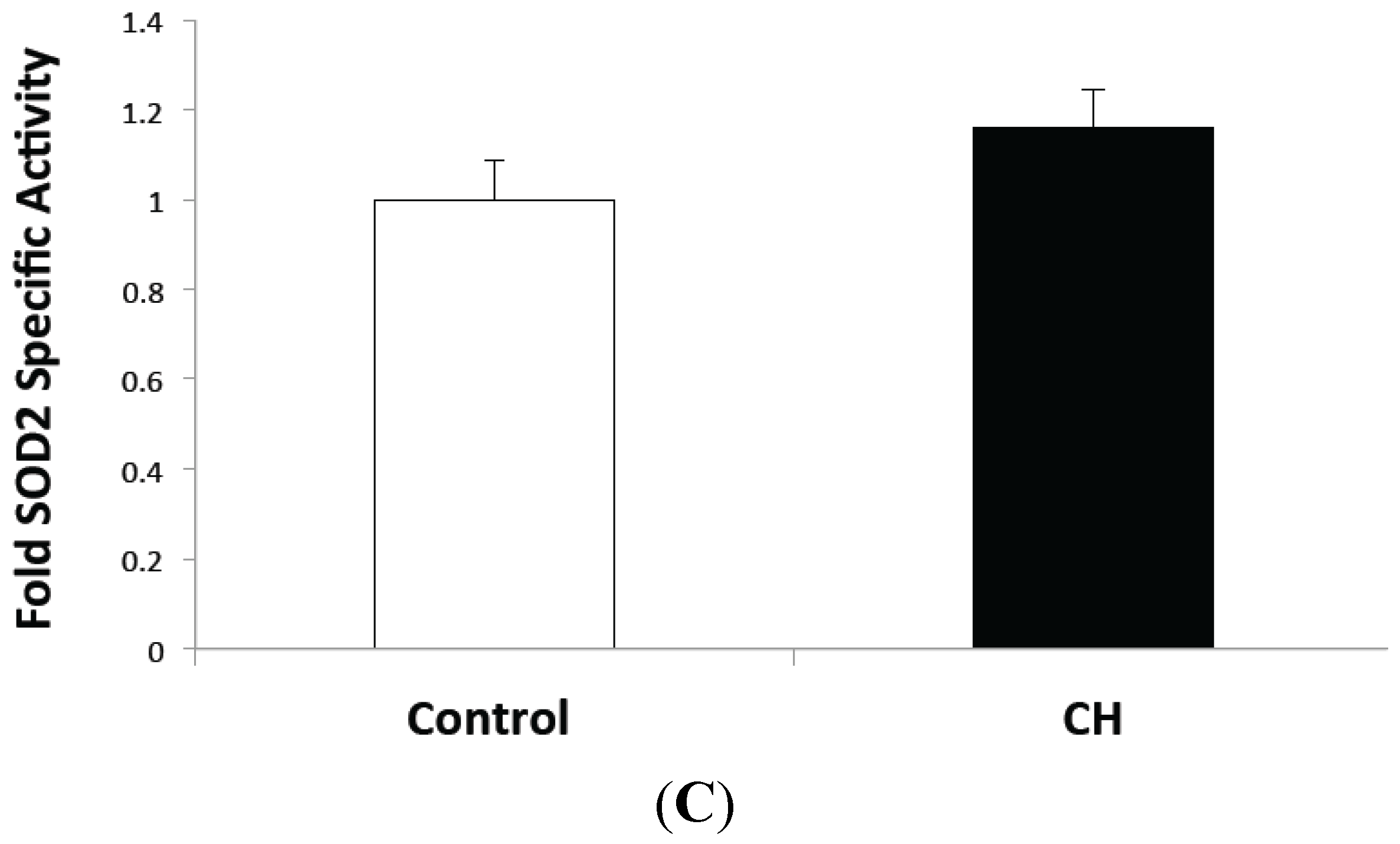
2.1. CH Exposed Lungs Have Increased SOD2 Expression but not Increased SOD2 Activity


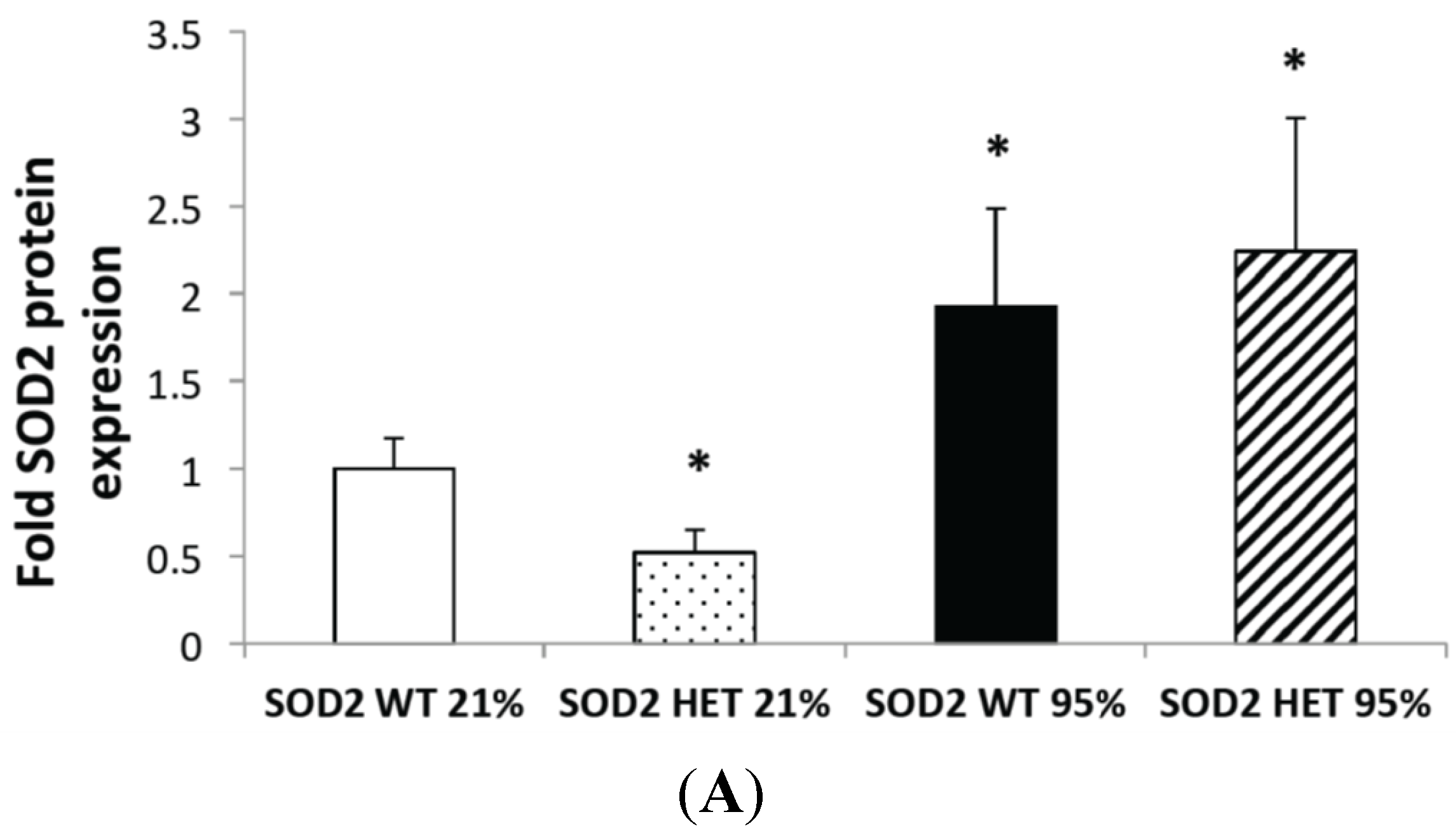
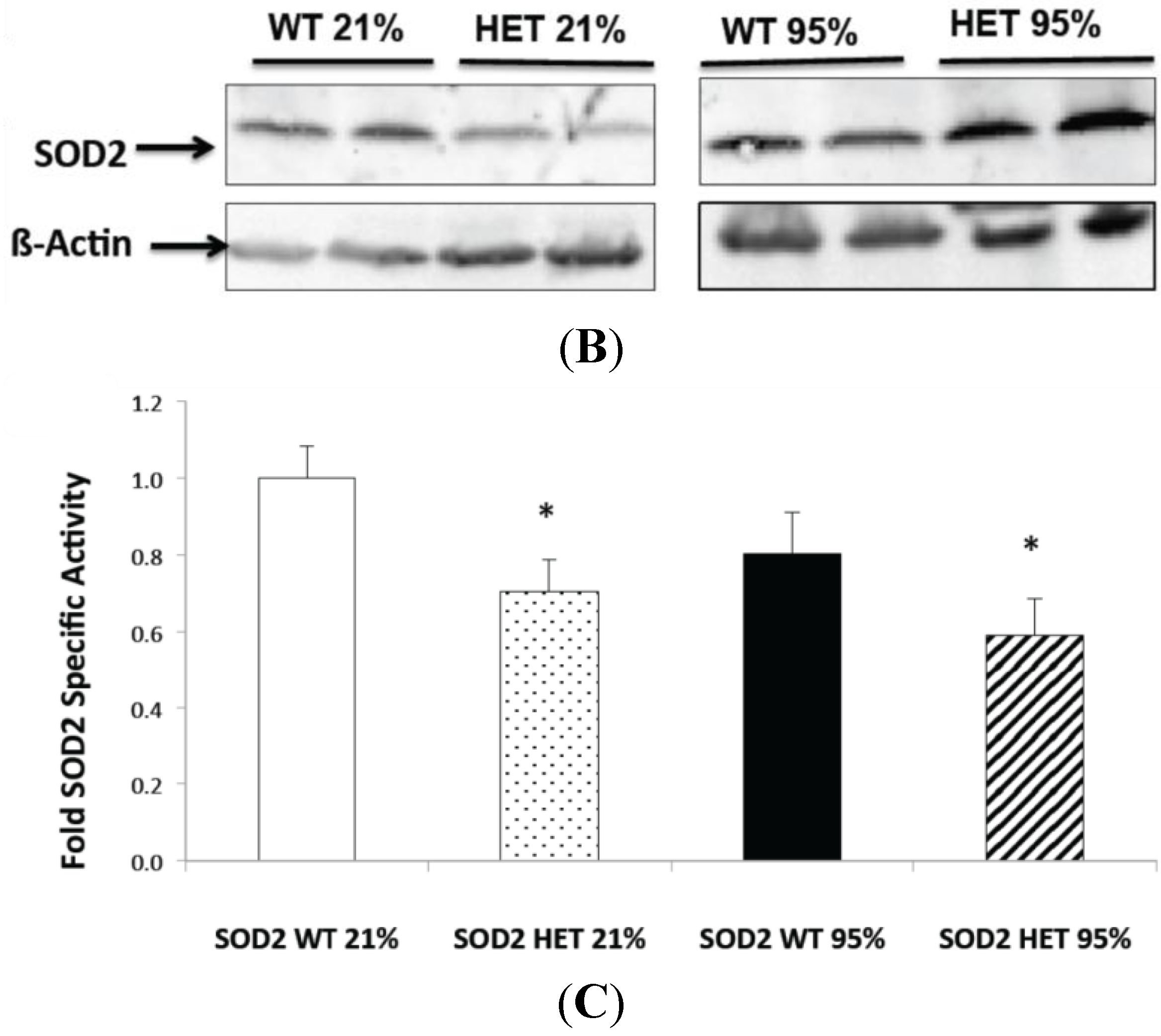
2.2. Hyperoxia Increased SOD2 Protein Expression in both SOD2+/+ and SOD2−/+ PASMC but not SOD2 Activity
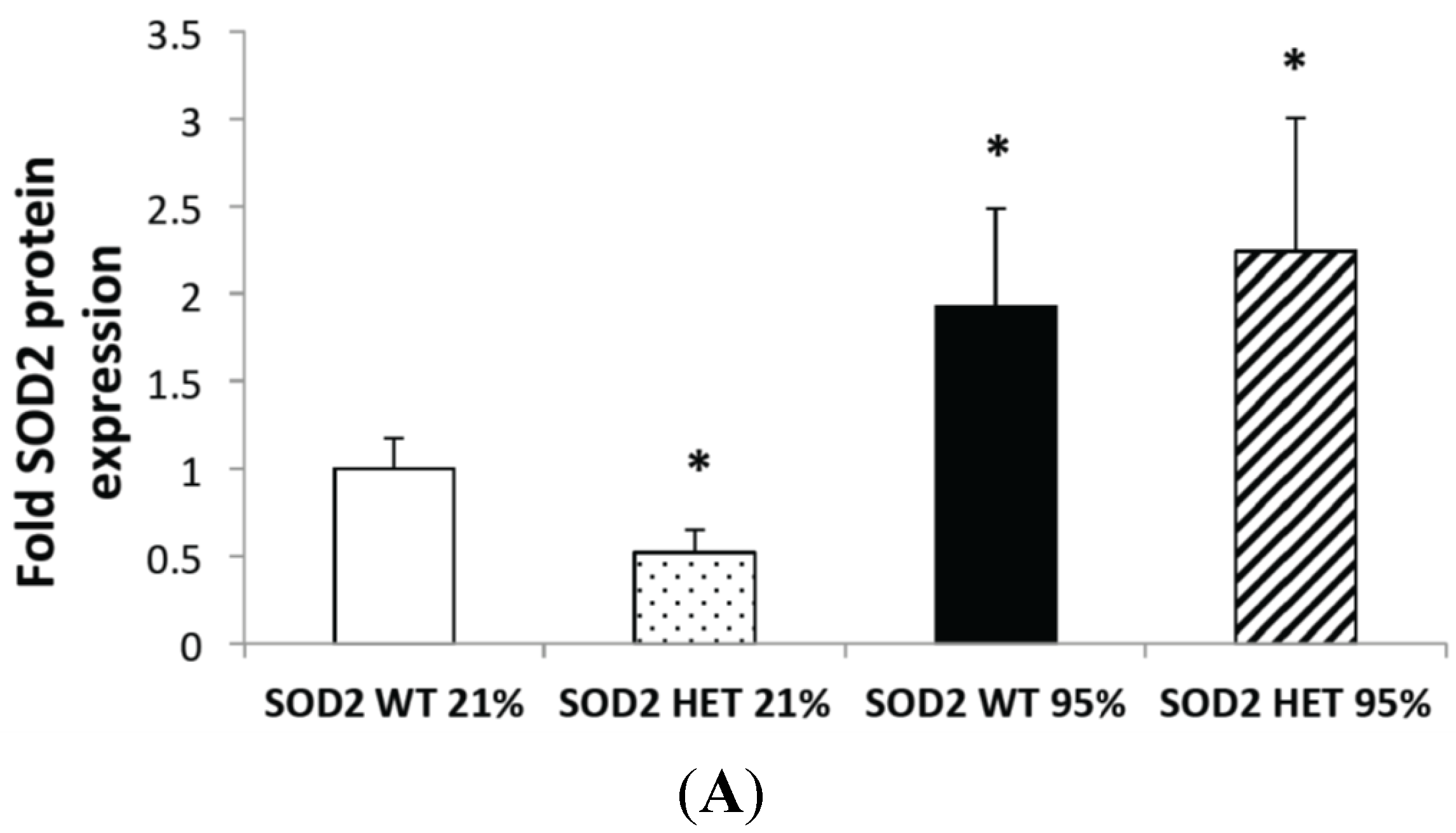

2.3. Mitochondrial Matrix Oxidative Stress Is Increased at Baseline and after Hyperoxia in SOD2−/+ PASMC Relative to Wild-Type Controls

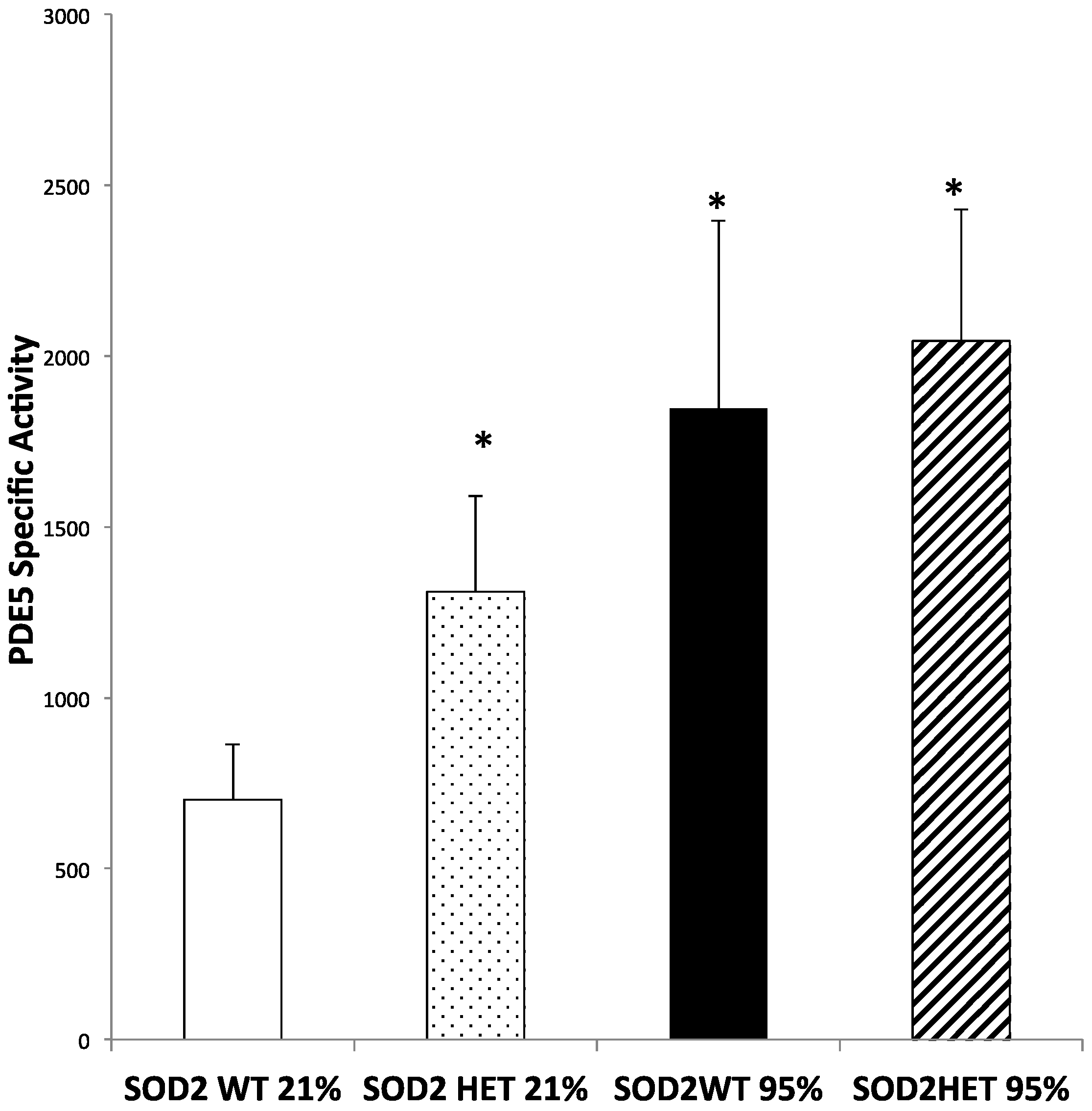
2.4. SOD2−/+ PASMC Have Increased PDE5 Activity at Baseline

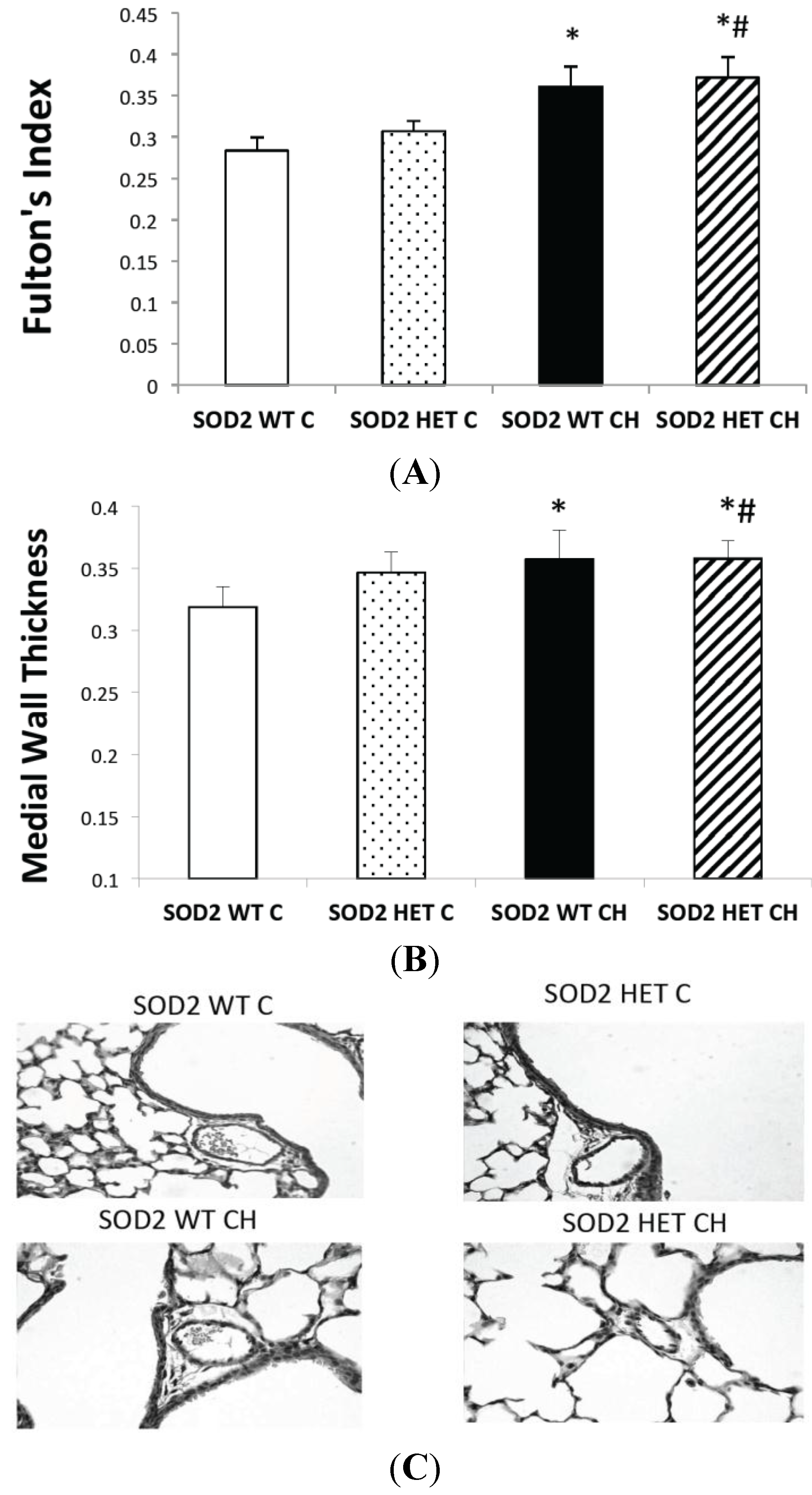
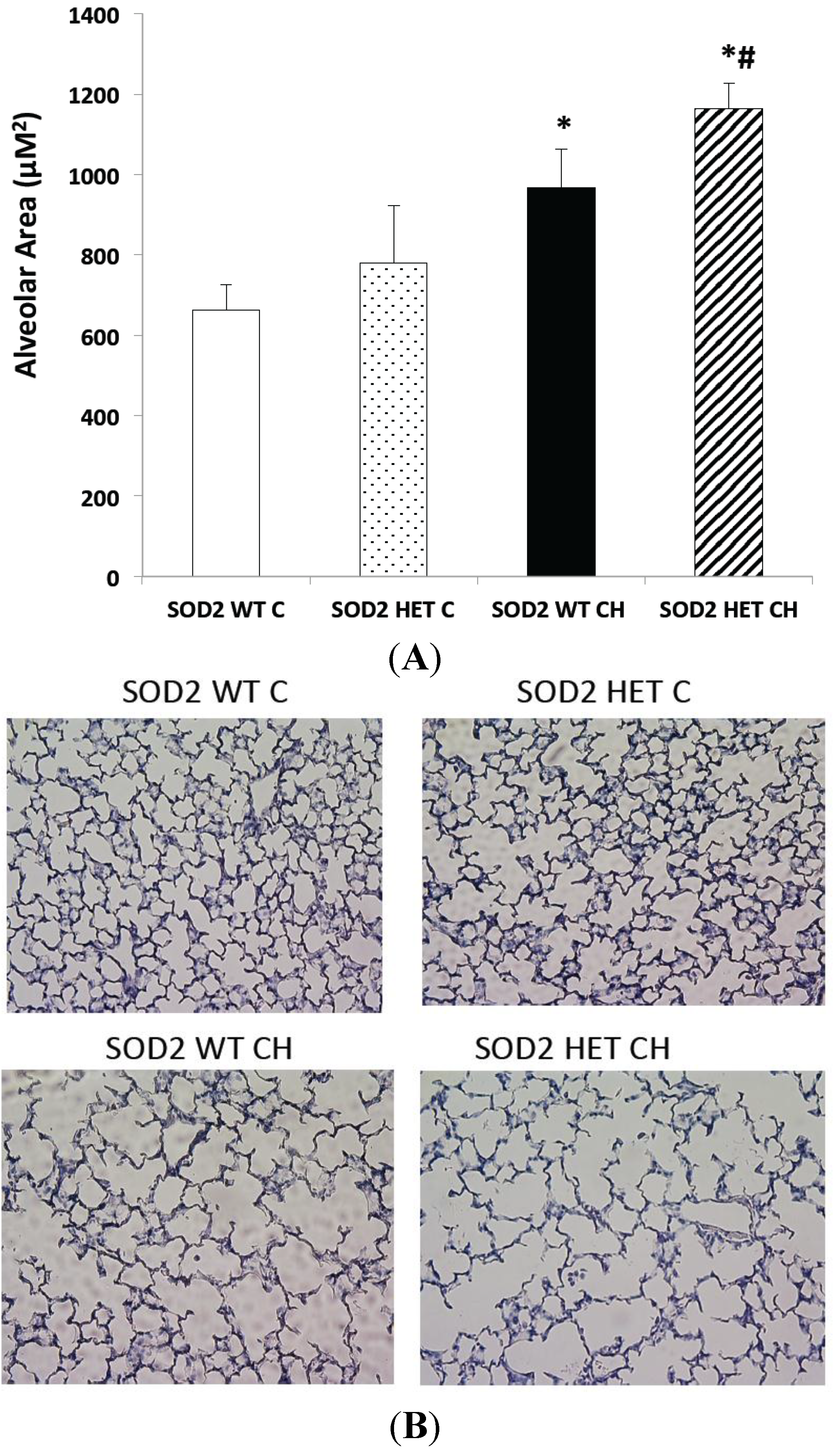
2.5. SOD2−/+ Mice Develop Equivalent Lung and Vascular Disease in Chronic Hyperoxia (CH) Relative to SOD2+/+ Littermates


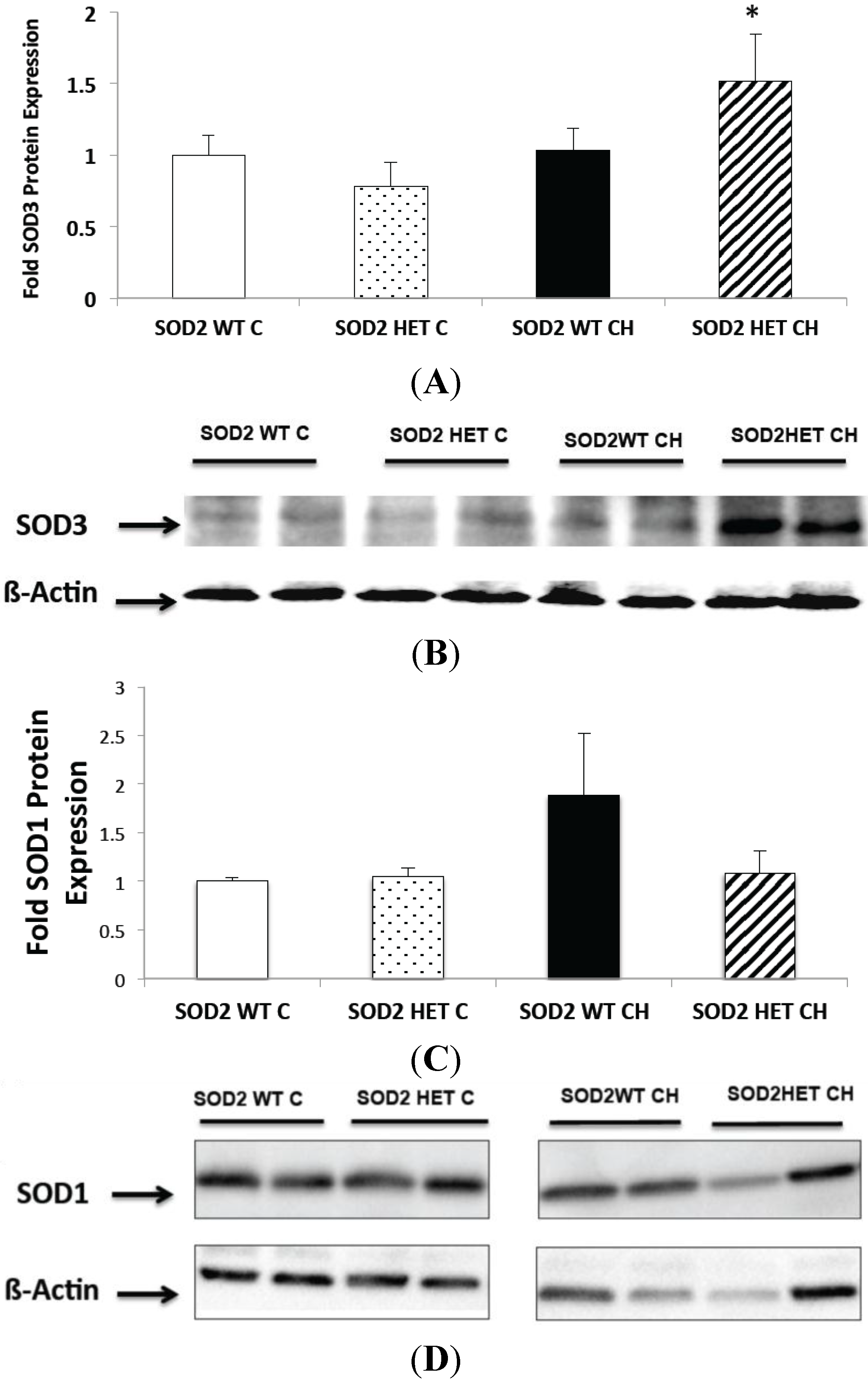
2.6. SOD2−/+ Mice Have Increased SOD3 Expression and Unchanged SOD1 Expression in Chronic Hyperoxia (CH)
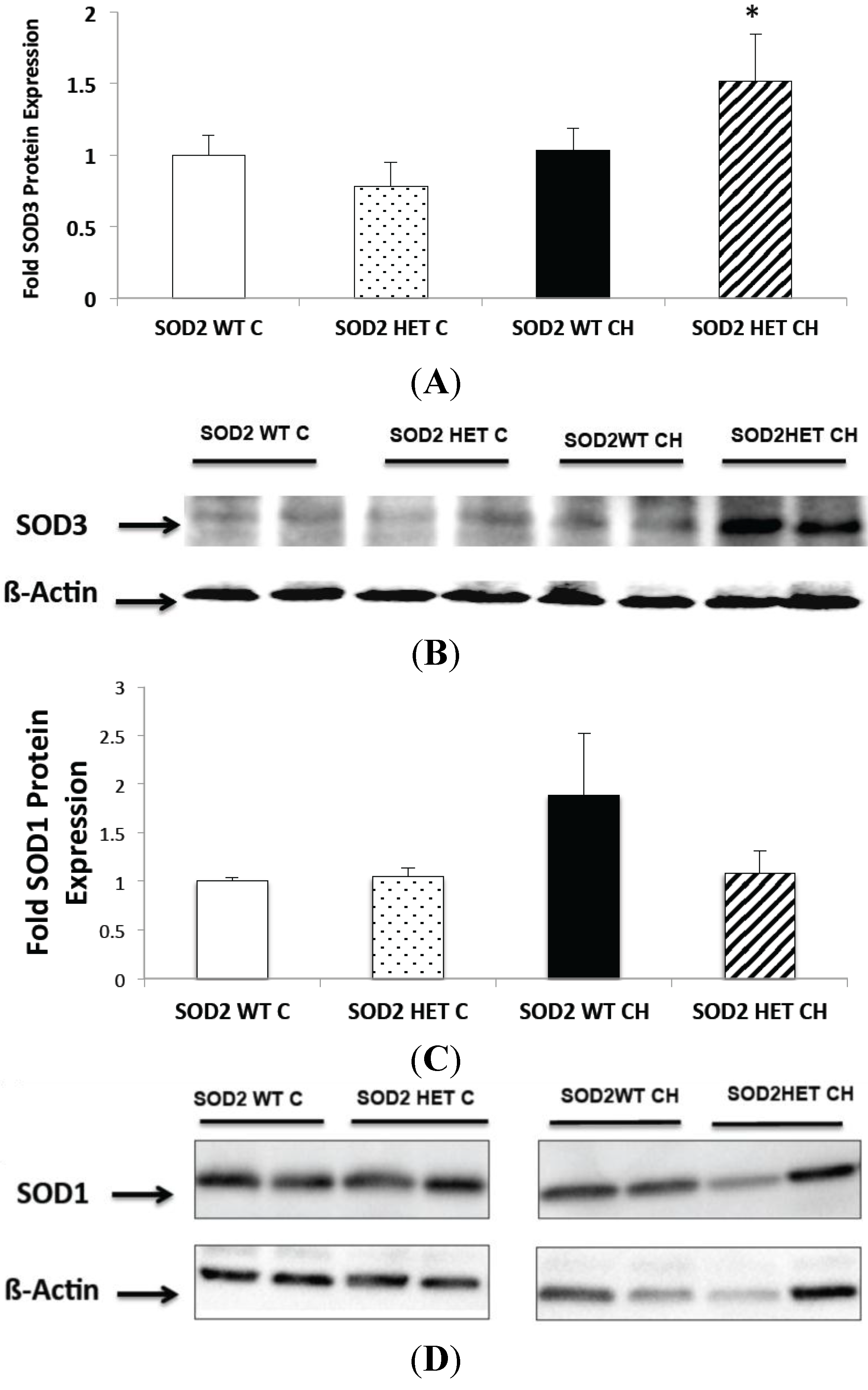
2.7. Discussion
3. Experimental Section
3.1. Animal Protocols
3.2. Isolation of Pulmonary Artery Smooth Muscle Cells (PASMC) and Exposure to Hyperoxia
3.3. Western Blot Analysis
3.4. SOD2 Activity Assay
3.5. Measurement of ROS
3.6. PDE5 Activity Assay
3.7. Measurement of Right Ventricular Hypertrophy (RVH)
3.8. Measurement of Medial Wall Thickness (MWT)
3.9. Measurement of Alveolar Area
3.10. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khemani, E.; McElhinney, D.B.; Rhein, L.; Andrade, O.; Lacro, R.V.; Thomas, K.C.; Mullen, M.P. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: Clinical features and outcomes in the surfactant era. Pediatrics 2007, 120, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Berkelhamer, S.K.; Mestan, K.K.; Steinhorn, R.H. Pulmonary hypertension in bronchopulmonary dysplasia. Semin. Perinatol. 2013, 37, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Check, J.; Gotteiner, N.; Liu, X.; Su, E.; Porta, N.; Steinhorn, R.; Mestan, K.K. Fetal growth restriction and pulmonary hypertension in premature infants with bronchopulmonary dysplasia. J. Perinatol. 2013, 33, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Padula, M.A.; Grover, T.R.; Brozanski, B.; Zaniletti, I.; Nelin, L.D.; Asselin, J.M.; Durand, D.J.; Short, B.L.; Pallotto, E.K.; Dykes, F.D.; et al. Therapeutic interventions and short-term outcomes for infants with severe bronchopulmonary dysplasia born at <32 weeks’ gestation. J. Perinatol. 2013, 33, 877–881. [Google Scholar]
- An, H.S.; Bae, E.J.; Kim, G.B.; Kwon, B.S.; Beak, J.S.; Kim, E.K.; Kim, H.S.; Choi, J.H.; Noh, C.I.; Yun, Y.S. Pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Korean Circ. J. 2010, 40, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.; Savani, R.C.; Lagatta, J.M.; Zaniletti, I.; Wadhawan, R.; Truog, W.; Grover, T.R.; Zhang, H.; Asselin, J.M.; Durand, D.J.; et al. Predicting death or tracheostomy placement in infants with severe bronchopulmonary dysplasia. J. Perinatol. 2014, 34, 543–548. [Google Scholar] [CrossRef]
- Farrow, K.N.; Wedgwood, S.; Lee, K.J.; Czech, L.; Gugino, S.F.; Lakshminrusimha, S.; Schumacker, P.T.; Steinhorn, R.H. Mitochondrial oxidant stress increases PED5 activity in persistent pulmonary hypertension of the newborn. Respir. Physiol. Neurobiol. 2010, 174, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.N.; Lee, K.J.; Perez, M.; Schriewer, J.M.; Wedgwood, S.; Lakshminrusimha, S.; Smith, C.L.; Steinhorn, R.H.; Schumacker, P.T. Brief hyperoxia increases mitochondrial oxidation and increases phosphodiesterase 5 activity in fetal pulmonary artery smooth muscle cells. Antioxid. Redox Signal. 2012, 17, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.N.; Groh, B.S.; Schumacker, P.T.; Lakshminrusimha, S.; Czech, L.; Gugino, S.F.; Russell, J.A.; Steinhorn, R.H. Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ. Res. 2008, 102, 226–233. [Google Scholar] [CrossRef]
- Lee, K.J.; Berkelhamer, S.K.; Kim, G.A.; Taylor, J.M.; O’Shea, K.M.; Steinhorn, R.H.; Farrow, K.N. Disrupted pulmonary artery cyclic guanosine monophosphate signaling in mice with hyperoxia-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 50, 369–378. [Google Scholar] [PubMed]
- Ookawara, T.; Imazeki, N.; Matsubara, O.; Kizaki, T.; Oh-Ishi, S.; Nakao, C.; Sato, Y.; Ohno, H. Tissue distribution of immunoreactive mouse extracellular superoxide dismutase. Am. J. Physiol. 1998, 275, C840–C847. [Google Scholar] [PubMed]
- Auten, R.L.; O’Reilly, M.A.; Oury, T.D.; Nozik-Grayck, E.; Whorton, M.H. Transgenic extracellular superoxide dismutase protects postnatal alveolar epithelial proliferation and development during hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L32–L40. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Parad, R.B.; Michele, T.; Allred, E.; Price, A.; Rosenfeld, W.; North American Recombinant Human CuZnSOD Study Group. Pulmonary outcome at 1 year corrected age in premature infants treated at birth with recombinant human cuzn superoxide dismutase. Pediatrics 2003, 111, 469–476. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. The definition and measurement of antioxidants in biological systems. Free Radic. Biol. Med. 1995, 18, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Wispe, J.R.; Warner, B.B.; Clark, J.C.; Dey, C.R.; Neuman, J.; Glasser, S.W.; Crapo, J.D.; Chang, L.Y.; Whitsett, J.A. Human Mn-superoxide dismutase in pulmonary epithelial cells of transgenic mice confers protection from oxygen injury. J. Biol. Chem. 1992, 267, 23937–23941. [Google Scholar] [PubMed]
- Guo, Z.M.; Yang, H.; Hamilton, M.L.; van Remmen, H.; Richardson, A. Effects of age and food restriction on oxidative DNA damage and antioxidant enzyme activities in the mouse aorta. Mech. Ageing Dev. 2001, 122, 1771–1786. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.M.; Helton, E.S.; Viera, L.; Ohman, T. Survival, lung injury, and lung protein nitration in heterozygous mnsod knockout mice in hyperoxia. Exp. Lung Res. 1999, 25, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, V.; Mervis, C.F.; Maxey, A.M.; Markham, N.E.; Abman, S.H. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: Implications for the pathogenesis of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1073–L1084. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Baveja, R.; Liang, O.D.; Fernandez-Gonzalez, A.; Lee, C.; Mitsialis, S.A.; Kourembanas, S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Amy, R.W.; Bowes, D.; Burri, P.H.; Haines, J.; Thurlbeck, W.M. Postnatal growth of the mouse lung. J. Anat. 1977, 124, 131–151. [Google Scholar] [PubMed]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Sam, F.; Kerstetter, D.L.; Pimental, D.R.; Mulukutla, S.; Tabaee, A.; Bristow, M.R.; Colucci, W.S.; Sawyer, D.B. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J. Card. Fail. 2005, 11, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Loch, T.; Vakhrusheva, O.; Piotrowska, I.; Ziolkowski, W.; Ebelt, H.; Braun, T.; Bober, E. Different extent of cardiac malfunction and resistance to oxidative stress in heterozygous and homozygous manganese-dependent superoxide dismutase-mutant mice. Cardiovasc. Res. 2009, 82, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D.; van Remmen, H.; Conrad, C.C.; Huang, T.T.; Epstein, C.J.; Richardson, A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J. Biol. Chem. 1998, 273, 28510–28515. [Google Scholar] [CrossRef] [PubMed]
- Kokoszka, J.E.; Coskun, P.; Esposito, L.A.; Wallace, D.C. Increased mitochondrial oxidative stress in the SOD2+/− mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. USA 2001, 98, 2278–2283. [Google Scholar] [CrossRef]
- Gong, Y.; Yi, M.; Fediuk, J.; Lizotte, P.P.; Dakshinamurti, S. Hypoxic neonatal pulmonary arterial myocytes are sensitized to ROS-generated 8-isoprostane. Free Radic. Biol. Med. 2010, 48, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Tsan, M.F.; White, J.E.; Caska, B.; Epstein, C.J.; Lee, C.Y. Susceptibility of heterozygous mnsod gene-knockout mice to oxygen toxicity. Am. J. Respir. Cell Mol. Biol. 1998, 19, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Czech, L.; Waypa, G.B.; Farrow, K.N. Isolation of pulmonary artery smooth muscle cells from neonatal mice. J. Vis. Exp. 2013, 80. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.N.; Lakshminrusimha, S.; Reda, W.J.; Wedgwood, S.; Czech, L.; Gugino, S.F.; Davis, J.M.; Russell, J.A.; Steinhorn, R.H. Superoxide dismutase restores enos expression and function in resistance pulmonary arteries from neonatal lambs with persistent pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L979–987. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Lakshminrusimha, S.; Czech, L.; Schumacker, P.T.; Steinhorn, R.H. Increased p22phox/Nox4 expression is involved in remodeling through hydrogen peroxide signaling in experimental persistent pulmonary hypertension of the newborn. Antioxid. Redox Signal. 2013, 18, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, A.; Perez, M.; Lee, K.J.; Taylor, J.M.; Farrow, K.N. SOD2 Activity Is not Impacted by Hyperoxia in Murine Neonatal Pulmonary Artery Smooth Muscle Cells and Mice. Int. J. Mol. Sci. 2015, 16, 6373-6390. https://doi.org/10.3390/ijms16036373
Gupta A, Perez M, Lee KJ, Taylor JM, Farrow KN. SOD2 Activity Is not Impacted by Hyperoxia in Murine Neonatal Pulmonary Artery Smooth Muscle Cells and Mice. International Journal of Molecular Sciences. 2015; 16(3):6373-6390. https://doi.org/10.3390/ijms16036373
Chicago/Turabian StyleGupta, Anita, Marta Perez, Keng Jin Lee, Joann M. Taylor, and Kathryn N. Farrow. 2015. "SOD2 Activity Is not Impacted by Hyperoxia in Murine Neonatal Pulmonary Artery Smooth Muscle Cells and Mice" International Journal of Molecular Sciences 16, no. 3: 6373-6390. https://doi.org/10.3390/ijms16036373
APA StyleGupta, A., Perez, M., Lee, K. J., Taylor, J. M., & Farrow, K. N. (2015). SOD2 Activity Is not Impacted by Hyperoxia in Murine Neonatal Pulmonary Artery Smooth Muscle Cells and Mice. International Journal of Molecular Sciences, 16(3), 6373-6390. https://doi.org/10.3390/ijms16036373