
Cholinergic Transactivation of the EGFR in HaCaT Keratinocytes Stimulates a Flotillin-1 Dependent MAPK-Mediated Transcriptional Response


Abstract
:
1. Introduction
2. Results and Discussion
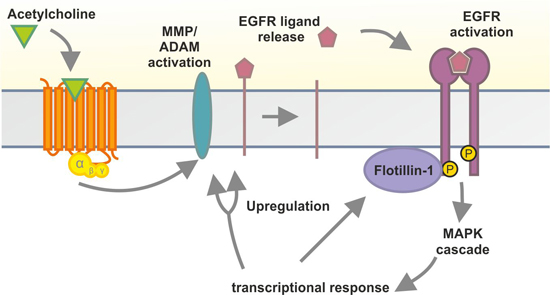
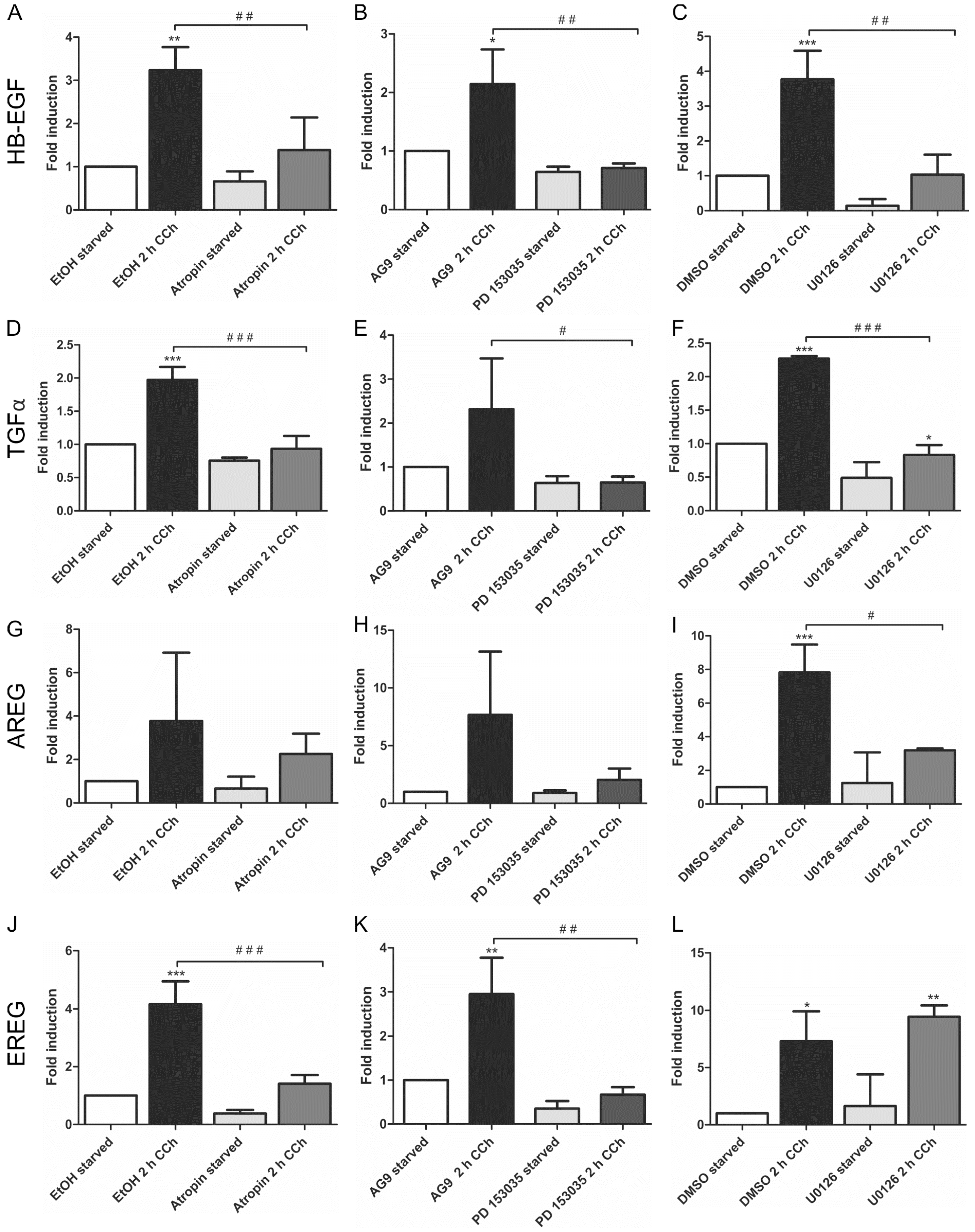
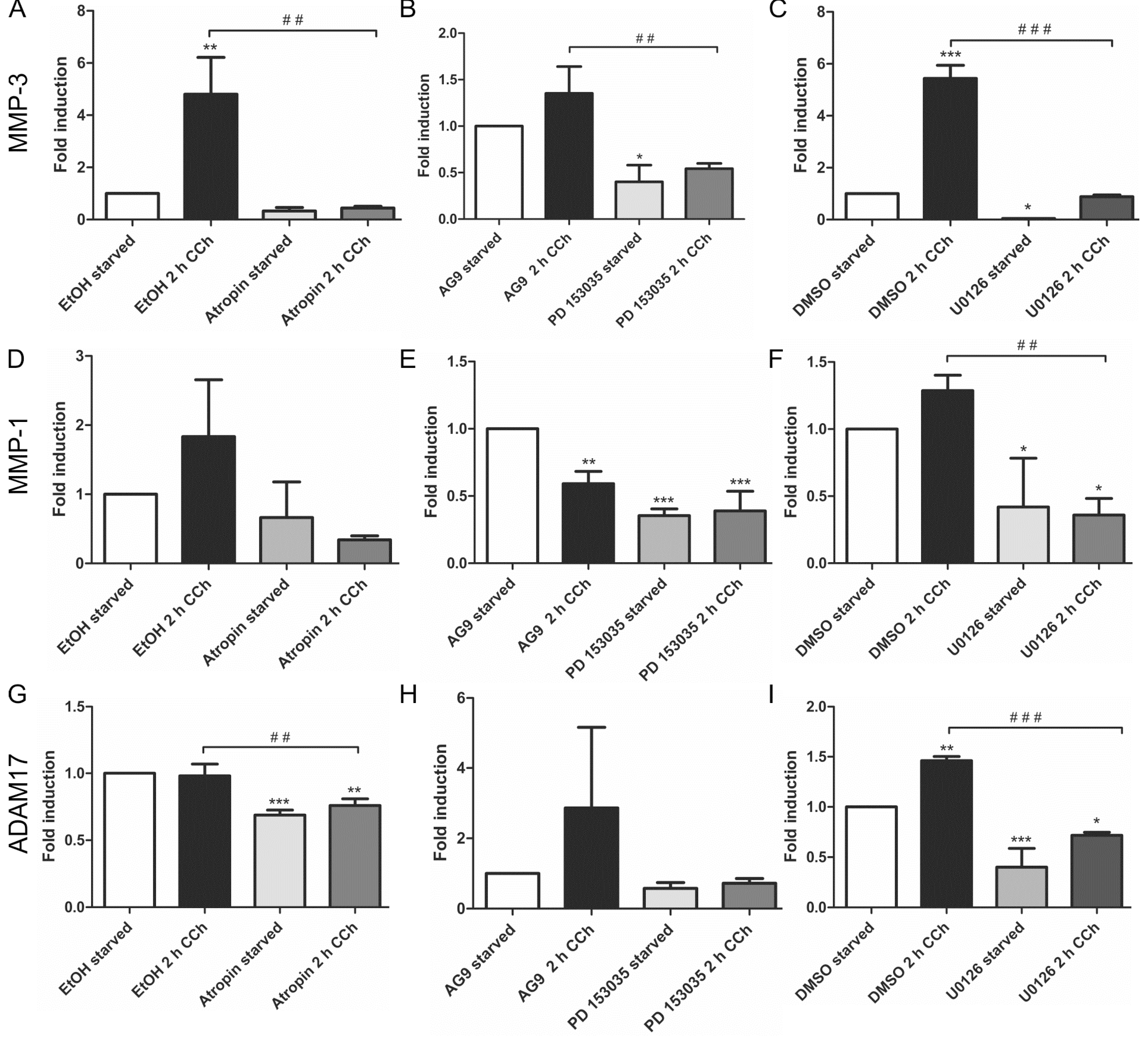
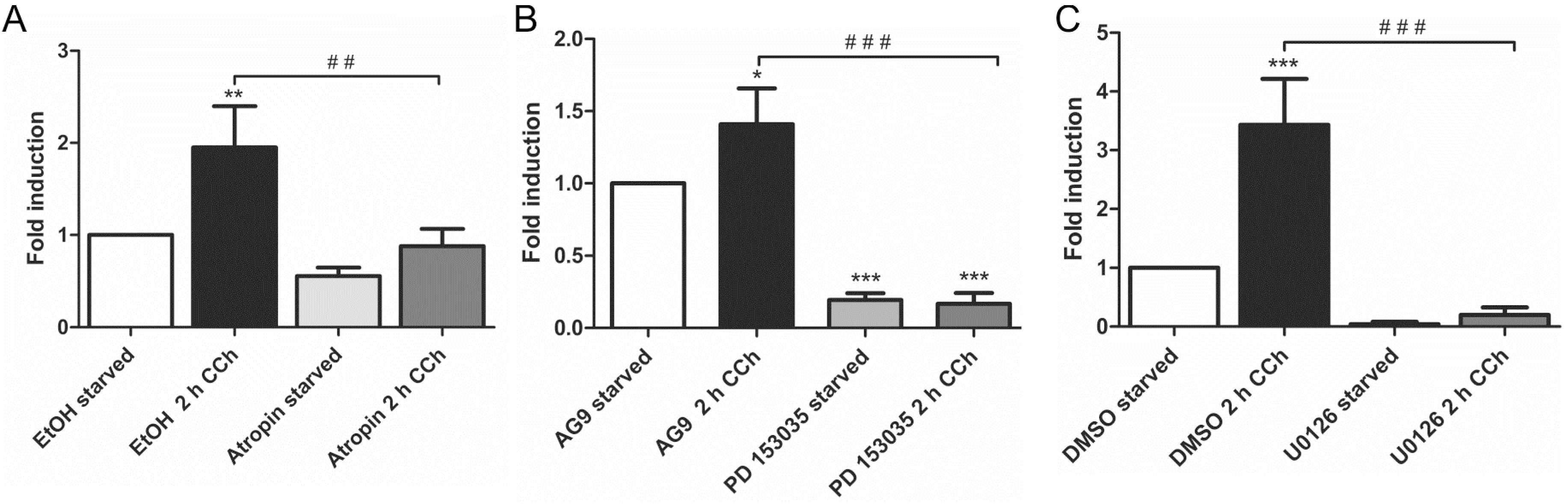
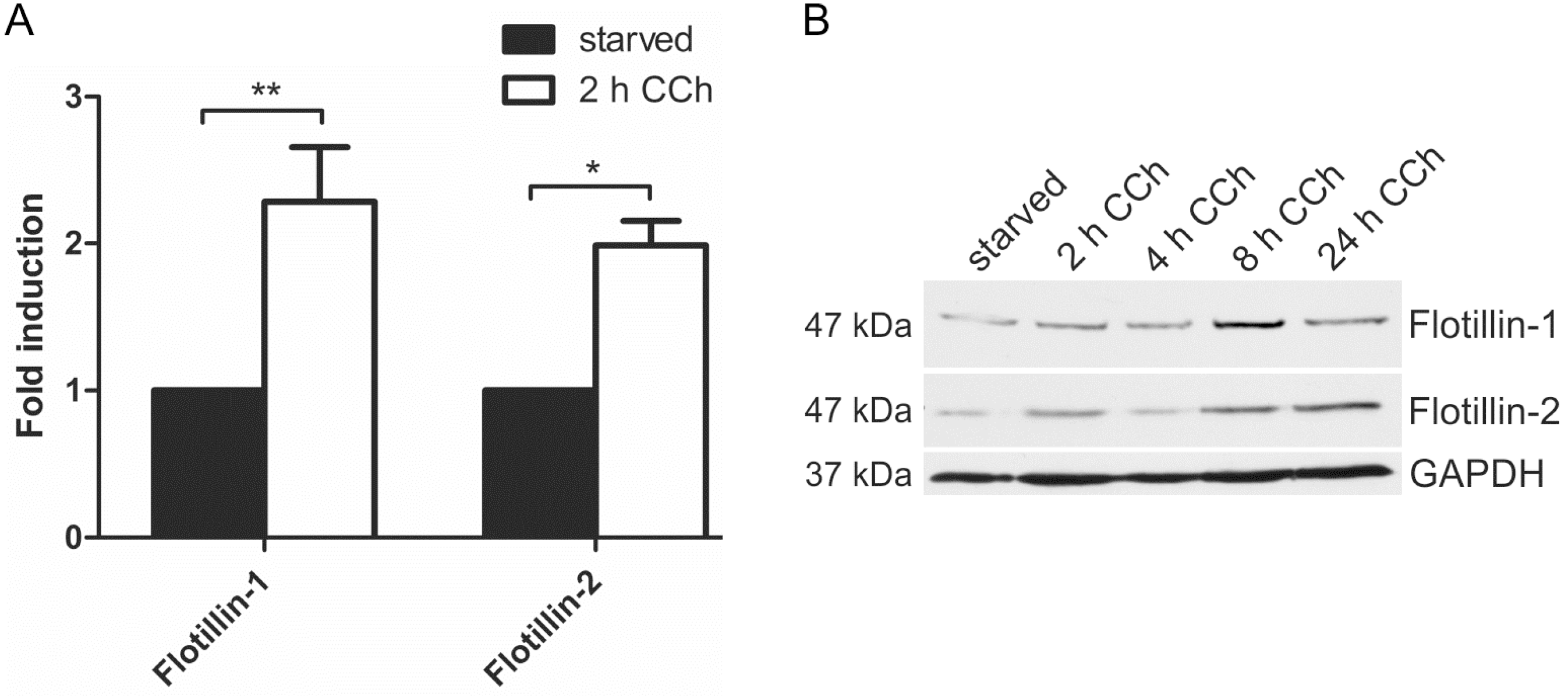
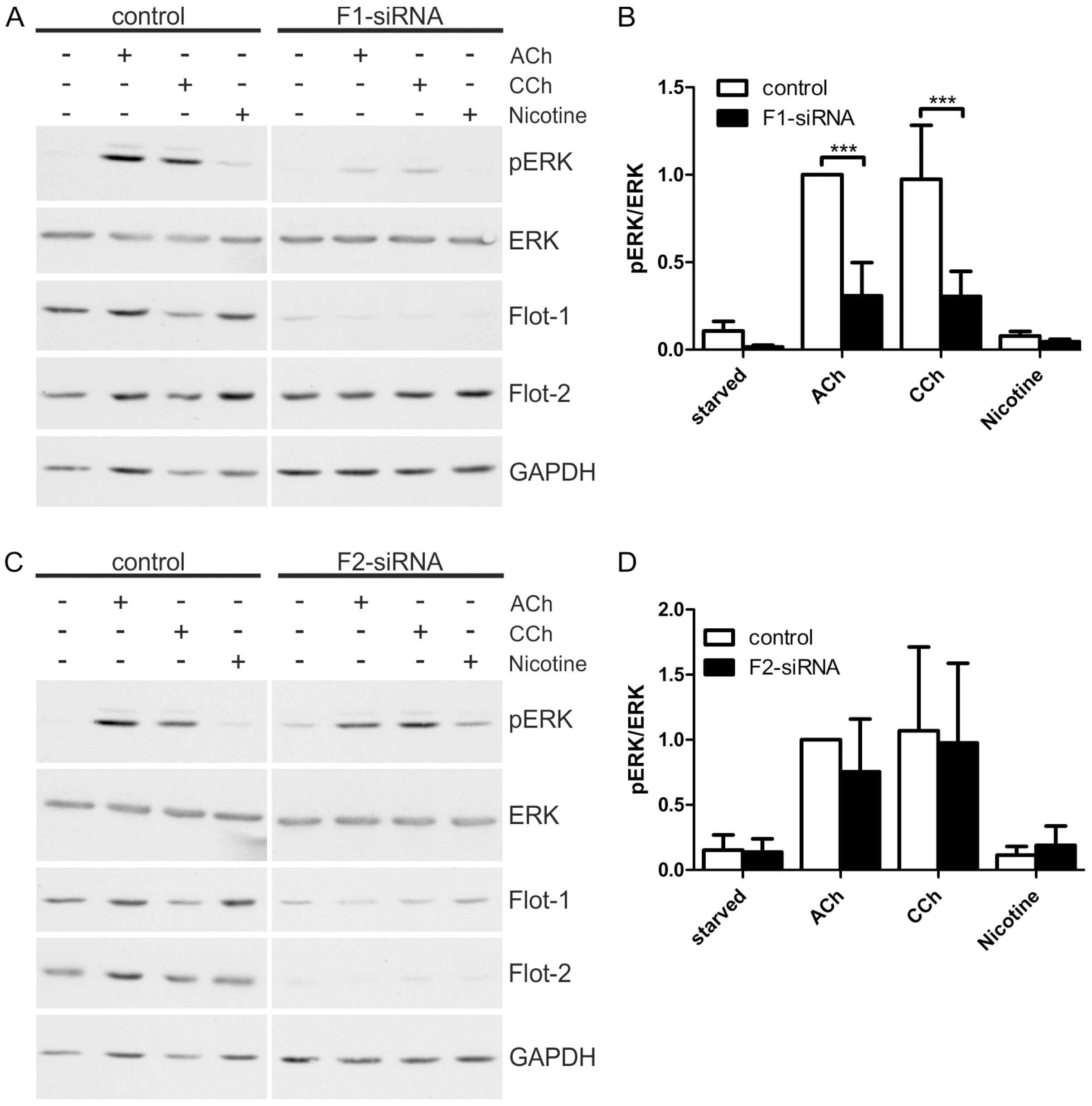
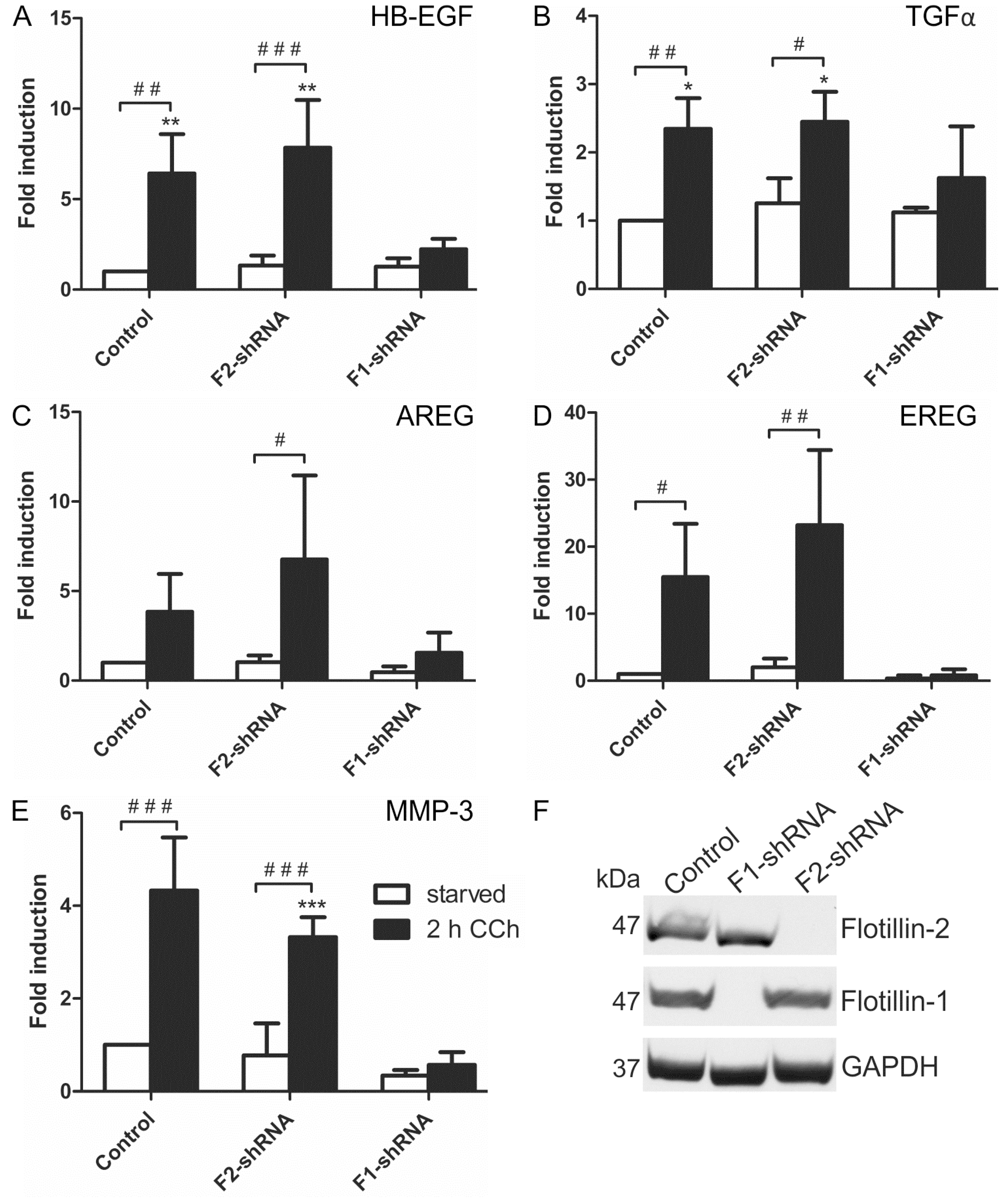
2.1. Results






2.2. Discussion
3. Experimental Section
3.1. Reagents and Antibodies
3.2. Cell Culture
3.3. Flotillin Knockdown by siRNA and shRNA
3.4. Cell Stimulation and Inhibitor Treatment
3.5. Cell Lysis, Gel Electrophoresis and Western Blot
3.6. RNA Isolation and RT-qPCR
3.7. Statistical Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Forward | Primer Reverse | TAnnealing (°C) |
|---|---|---|---|
| cFos | TGGTGAAGACCGTGTCAGGAG | TGATCTGTCTCCGCTTGGAGTG | 60 |
| Dusp1 | GGAGGACAACCAGGCAGAC | AGGTAAGCAAGGCAGATGGTGG | 60 |
| Egr1 | TTCAACCCTCAGGCGGACAC | GTCTCCACCAGCACCTTCTCGT | 60 |
| HPRT | GCAGTCCCAGGGTGCGTG | GGCCTCCCATCTCCTTCAT | 60 |
| Ywhaz | AGGTTGCCGCTGGTGATGAC | GGCCAGACCCAGTCTGATAGGA | 60 |
| Rpl13a | CCTGGAGGAGAAGAGGAAAGAGA | TTGAGGACCTCTGTGTATTTGTCAA | 60 |
| BTC | GCGGAAAGGCCACTTCTCTAGG | TCATCACAGACACAGGAGGGCG | 60 |
| EGF | GCAGCTTCAGGACCACAACCA | AAACCATTCCCATCTGCTGGCT | 60 |
| EPGN | ACTCCAGAGGCTGACACAGGAC | GGAGTTTCGCTCTTGTCACCCA | 60 |
| HB-EGF | GTCTGTCTGCTGGTCATCGTGG | CCCAGCCGATTCCTTGAGCA | 60 |
| EREG | GGTTTCCATCTTCTACAGGCA | TGTCTTCTGTCTGAACTAAAGCTG | 60 |
| AREG | GATACTCGGCTCAGGCCATTATGC | TCAAATCCATCAGCACTGTGGTCC | 60 |
| TGFα | GCTGATACACTGCTGCCAGGTC | AGCAAGCGGTTCTTCCCTTCAG | 60 |
| ADAM17 | TCGAGGGTGGATGAAGGAGAAGAG | GGACTGTTCCTGTCACTGCACTG | 62 |
| ADAM10 | TCCACAGCCCATTCAGCAA | GCGTCTCATGTGTCCCATTTG | 60 |
| ADAM8 | CACCAAAGCAGGTCATCAAGCCA | GCAACCTTTGGGCCAACAGCAC | 62 |
| MMP-1 | CTGCCAAATGGGCTTGAAGCTGC | AGTTCTAGGGAAGCCAAAGGAGCT | 62 |
| MMP-2 | CCTGACATTGACCTTGGCACCG | GTCCGCCAAATGAACCGGTCCT | 62 |
| MMP-3 | ATTCAGTCCCTCTATGGACCTCCC | ACAGCATCAAAGGACAAAGCAGGA | 58 |
| MMP-7 | TGGGAACAGGCTCAGGACTATCTC | TGACGCGGGAGTTTAACATTCCAG | 62 |
| MMP-9 | TTCTTCTCTGGGCGCCAGGT | CCGCTGAACAGCAGCATCTTCC | 58 |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grando, S.A.; Pittelkow, M.R.; Schallreuter, K.U. Adrenergic and cholinergic control in the biology of epidermis: Physiological and clinical significance. J. Investig. Dermatol. 2006, 126, 1948–1965. [Google Scholar] [CrossRef] [PubMed]
- Kummer, W.; Lips, K.S.; Pfeil, U. The epithelial cholinergic system of the airways. Histochem. Cell Biol. 2008, 130, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Ockenga, W.; Kühne, S.; Bocksberger, S.; Banning, A.; Tikkanen, R. Non-neuronal functions of the M2 muscarinic acetylcholine receptor. Genes 2013, 4, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Wallasch, C.; Lankenau, A.; Herrlich, A.; Ullrich, A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997, 16, 7032–7044. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Zimniak, P.; Raufman, J.P. Transactivation of the epidermal growth factor receptor mediates cholinergic agonist-induced proliferation of H508 human colon cancer cells. Cancer Res. 2003, 63, 6744–6750. [Google Scholar] [PubMed]
- Keely, S.J.; Calandrella, S.O.; Barrett, K.E. Carbachol-stimulated transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T(84) cells is mediated by intracellular Ca(2+), PYK-2, and p60(SRC). J. Biol. Chem. 2000, 275, 12619–12625. [Google Scholar] [CrossRef] [PubMed]
- McCole, D.F.; Keely, S.J.; Coffey, R.J.; Barrett, K.E. Transactivation of the epidermal growth factor receptor in colonic epithelial cells by carbachol requires extracellular release of transforming growth factor-alpha. J. Biol. Chem. 2002, 277, 42603–42612. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, C. EGF receptor activation by GPCRs: An universal pathway reveals different versions. Mol. Cell. Endocrinol. 2011, 331, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Jaldety, Y.; Glick, Y.; Orr-Urtreger, A.; Ickowicz, D.; Gerber, D.; Breitbart, H. Sperm epidermal growth factor receptor (EGFR) mediates alpha7 acetylcholine receptor (AChR) activation to promote fertilization. J. Biol. Chem. 2012, 287, 22328–22340. [Google Scholar] [CrossRef] [PubMed]
- Meister, M.; Tomasovic, A.; Banning, A.; Tikkanen, R. Mitogen-activated protein (MAP) kinase scaffolding proteins: A recount. Int. J. Mol. Sci. 2013, 14, 4854–4884. [Google Scholar] [CrossRef] [PubMed]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [PubMed]
- Wallasch, C.; Crabtree, J.E.; Bevec, D.; Robinson, P.A.; Wagner, H.; Ullrich, A. Helicobacter pylori-stimulated EGF receptor transactivation requires metalloprotease cleavage of HB-EGF. Biochem. Biophys. Res. Commun. 2002, 295, 695–701. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Hannan, R.D.; Thomas, W.G. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013, 280, 5258–5268. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Zwick, E.; Prenzel, N.; Leserer, M.; Ullrich, A. Cell communication networks: Epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene 2001, 20, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Leserer, M.; Gschwind, A.; Ullrich, A. Epidermal growth factor receptor signal transactivation. IUBMB Life 2000, 49, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Hawes, B.E.; Luttrell, L.M.; van Biesen, T.; Lefkowitz, R.J. Phosphatidylinositol 3-kinase is an early intermediate in the G beta gamma-mediated mitogen-activated protein kinase signaling pathway. J. Biol. Chem. 1996, 271, 12133–12136. [Google Scholar] [CrossRef] [PubMed]
- Hawes, B.E.; van Biesen, T.; Koch, W.J.; Luttrell, L.M.; Lefkowitz, R.J. Distinct pathways of Gi- and Gq-mediated mitogen-activated protein kinase activation. J. Biol. Chem. 1995, 270, 17148–17153. [Google Scholar] [CrossRef] [PubMed]
- Keely, S.J.; Uribe, J.M.; Barrett, K.E. Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T 84 cells. J. Biol. Chem. 1998, 273, 27111–27117. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Hawes, B.E.; van Biesen, T.; Luttrell, D.K.; Lansing, T.J.; Lefkowitz, R.J. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinases. J. Biol. Chem. 1996, 271, 19443–19450. [Google Scholar] [CrossRef] [PubMed]
- Coffey, R.J., Jr.; Derynck, R.; Wilcox, J.N.; Bringman, T.S.; Goustin, A.S.; Moses, H.L.; Pittelkow, M.R. Production and auto-induction of transforming growth factor-alpha in human keratinocytes. Nature 1987, 328, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Cook, P.W.; Mattox, P.A.; Keeble, W.W.; Pittelkow, M.R.; Plowman, G.D.; Shoyab, M.; Adelman, J.P.; Shipley, G.D. A heparin sulfate-regulated human keratinocyte autocrine factor is similar or identical to amphiregulin. Mol. Cell. Biol. 1991, 11, 2547–2557. [Google Scholar] [PubMed]
- Hashimoto, K.; Higashiyama, S.; Asada, H.; Hashimura, E.; Kobayashi, T.; Sudo, K.; Nakagawa, T.; Damm, D.; Yoshikawa, K.; Taniguchi, N. Heparin-binding epidermal growth factor-like growth factor is an autocrine growth factor for human keratinocytes. J. Biol. Chem. 1994, 269, 20060–20066. [Google Scholar] [PubMed]
- Shirakata, Y.; Komurasaki, T.; Toyoda, H.; Hanakawa, Y.; Yamasaki, K.; Tokumaru, S.; Sayama, K.; Hashimoto, K. Epiregulin, a novel member of the epidermal growth factor family, is an autocrine growth factor in normal human keratinocytes. J. Biol. Chem. 2000, 275, 5748–5753. [Google Scholar] [CrossRef] [PubMed]
- Pastore, S.; Mascia, F.; Mariani, V.; Girolomoni, G. The epidermal growth factor receptor system in skin repair and inflammation. J. Investig. Dermatol. 2008, 128, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Ockenga, W.; Kühne, S.; Bocksberger, S.; Banning, A.; Tikkanen, R. Epidermal growth factor receptor transactivation is required for mitogen-activated protein kinase activation by muscarinic acetylcholine receptors in HaCaT keratinocytes. Int. J. Mol. Sci. 2014, 15, 21433–21454. [Google Scholar] [CrossRef] [PubMed]
- Amaddii, M.; Meister, M.; Banning, A.; Tomasovic, A.; Mooz, J.; Rajalingam, K.; Tikkanen, R. Flotillin-1/reggie-2 protein plays dual role in activation of receptor-tyrosine kinase/mitogen-activated protein kinase signaling. J. Biol. Chem. 2012, 287, 7265–7278. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Kurrle, N.; Meister, M.; Tikkanen, R. Flotillins in receptor tyrosine kinase signaling and cancer. Cells 2014, 3, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Regenbrecht, C.R.; Tikkanen, R. Increased activity of mitogen activated protein kinase pathway in flotillin-2 knockout mouse model. Cell Signal. 2014, 26, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Ockenga, W.; Finger, F.; Siebrasse, P.; Tikkanen, R. Transcriptional regulation of flotillins by the extracellularly regulated kinases and retinoid X receptor complexes. PLoS One 2012, 7, e45514. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Cheng, K.; Shant, J.; Raufman, J.P. Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G755–G763. [Google Scholar] [CrossRef] [PubMed]
- Cury, P.R.; de Araujo, V.C.; Canavez, F.; Furuse, C.; Leite, K.R.; de Araujo, N.S. The effect of epidermal growth factor on matrix metalloproteinases and tissue inhibitors of metalloproteinase gene expression in cultured human gingival fibroblasts. Arch. Oral Biol. 2007, 52, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Domeij, H.; Yucel-Lindberg, T.; Modeer, T. Signal pathways involved in the production of MMP-1 and MMP-3 in human gingival fibroblasts. Eur. J. Oral Sci. 2002, 110, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Kajanne, R.; Miettinen, P.; Mehlem, A.; Leivonen, S.K.; Birrer, M.; Foschi, M.; Kahari, V.M.; Leppa, S. EGF-R regulates MMP function in fibroblasts through MAPK and AP-1 pathways. J. Cell Physiol. 2007, 212, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Nutt, J.E.; Durkan, G.C.; Mellon, J.K.; Lunec, J. Matrix metalloproteinases (MMPs) in bladder cancer: The induction of MMP9 by epidermal growth factor and its detection in urine. BJU Int. 2003, 91, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.P.; Dempsey, P.J.; Dunbar, A.J. Control of ErbB signaling through metalloprotease mediated ectodomain shedding of EGF-like factors. Growth Factors 2006, 24, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R. The magnificent seven: Epidermal growth factor receptor ligands. Semin. Cell Dev. Biol. 2014, 28, 1. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Wolf, E. The epidermal growth factor receptor ligands at a glance. J. Cell Physiol. 2009, 218, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Grandis, J.R. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front. Biosci. 2008, 13, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Kalyankrishna, S.; Grandis, J.R. Epidermal growth factor receptor biology in head and neck cancer. J. Clin. Oncol. 2006, 24, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Hart, S.; Fischer, O.M.; Ullrich, A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 2003, 22, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Schafer, B.; Marg, B.; Gschwind, A.; Ullrich, A. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J. Biol. Chem. 2004, 279, 47929–47938. [Google Scholar] [CrossRef] [PubMed]
- Hazarika, P.; McCarty, M.F.; Prieto, V.G.; George, S.; Babu, D.; Koul, D.; Bar-Eli, M.; Duvic, M. Up-regulation of Flotillin-2 is associated with melanoma progression and modulates expression of the thrombin receptor protease activated receptor 1. Cancer Res. 2004, 64, 7361–7369. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, Y.; Nishii, H.; Takahashi, T.; Yamauchi, J.; Mizuno, N.; Tago, K.; Itoh, H. The lipid raft proteins flotillins/reggies interact with Galphaq and are involved in Gq-mediated p38 mitogen-activated protein kinase activation through tyrosine kinase. Cell Signal. 2007, 19, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A. Connections of nicotine to cancer. Nat. Rev. Cancer 2014, 14, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Babuke, T.; Ruonala, M.; Meister, M.; Amaddii, M.; Genzler, C.; Esposito, A.; Tikkanen, R. Hetero-oligomerization of reggie-1/flotillin-2 and reggie-2/flotillin-1 is required for their endocytosis. Cell Signal. 2009, 21, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Kurrle, N.; Völlner, F.; Eming, R.; Hertl, M.; Banning, A.; Tikkanen, R. Flotillins directly interact with gamma-catenin and regulate epithelial cell–cell adhesion. PLoS One 2013, 8, e84393. [Google Scholar] [CrossRef] [PubMed]
- Kurrle, N.; Ockenga, W.; Meister, M.; Völlner, F.; Kühne, S.; John, B.A.; Banning, A.; Tikkanen, R. Phosphatidylinositol 3-Kinase dependent upregulation of the epidermal growth factor receptor upon Flotillin-1 depletion in breast cancer cells. BMC Cancer 2013, 13, 575. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kühne, S.; Ockenga, W.; Banning, A.; Tikkanen, R. Cholinergic Transactivation of the EGFR in HaCaT Keratinocytes Stimulates a Flotillin-1 Dependent MAPK-Mediated Transcriptional Response. Int. J. Mol. Sci. 2015, 16, 6447-6463. https://doi.org/10.3390/ijms16036447
Kühne S, Ockenga W, Banning A, Tikkanen R. Cholinergic Transactivation of the EGFR in HaCaT Keratinocytes Stimulates a Flotillin-1 Dependent MAPK-Mediated Transcriptional Response. International Journal of Molecular Sciences. 2015; 16(3):6447-6463. https://doi.org/10.3390/ijms16036447
Chicago/Turabian StyleKühne, Sina, Wymke Ockenga, Antje Banning, and Ritva Tikkanen. 2015. "Cholinergic Transactivation of the EGFR in HaCaT Keratinocytes Stimulates a Flotillin-1 Dependent MAPK-Mediated Transcriptional Response" International Journal of Molecular Sciences 16, no. 3: 6447-6463. https://doi.org/10.3390/ijms16036447