1. Introduction
B. subtilis, a type of Gram-positive bacterium, has been used as a cell factory for many enzymes, including proteases, amylases, and lipases [
1,
2,
3]. Some subspecies produce antifungal lipopeptides (surfactin, iturin, and fengycin) that are widely applied for the control of plant pathogenic fungi in agriculture.
B. subtilis ZK, a wild-type
B. subtilis with high economic value, has been a workhorse for producing iturin A at a high level [
4]. In order to study the high yield mechanism and further improve the iturin A production, it is desirable to perform molecular genetics studies in
B. subtilis ZK. However, the fact that the genetic manipulation systems of
B. subtilis ZK, such as the transformation system, gene deletion and mutation library, have not been established retards molecular genetic studies of this organism. The present work aimed at developing an efficient transformation method for the
B. subtilis ZK in order to carry out molecular genetics studies.
There are five transformation systems available that have been developed to introduce exogenous DNA into
B. subtilis, such as phage transduction [
5], protoplast [
6], natural transformation [
7], recombinant method [
8], and electroporation [
9]. The first two methods are important methods for introducing DNA into
Bacillus [
10]. Unfortunately, these two methods have not been wildly used mainly due to heavy workload and time-consuming experimental processes [
11]. Natural transformation and recombinant methods are usually used for introducing DNA into
B. subtilis and lead to high transformation efficiency for some
B. subtilis [
7,
8]. However, the transformation efficiency of these two methods is closely related to some genes, such as quorum sensing components, and competence master regulator
comK. The divergent structures of those genes lead to a huge difference in the transformation efficiency [
12,
13]. Hence, the natural transformation and recombinant method are strain-specific. In contrast to other methods, electroporation is a common, time-saving transformation method and has been widely used for
Bacillus [
9,
10,
11,
14,
15]. Although the electroporation system for
B. subtilis has been developed for many years, there is still no standard and universal protocol for all
B. subtilis. We carried out an efficient protocol according to the reported literature [
9]. Unfortunately, relatively low efficiency was obtained (about 1.0 × 10
3 transformants/μg DNA) in wild-type
B. subtilis ZK and the knockout of a gene was not to be performed, because the electroporation efficiency (1.0 × 10
3 transformants/μg DNA) was insufficient and the frequency of a foreign plasmid integrating into genome was about 1.0 × 10
−4–1.0 × 10
−6 per plasmid when the homologous region was less than 1 kb [
16,
17,
18,
19].
Many factors affect electroporation efficiency including growth medium [
11], growth phase [
14], electric field [
20], weakening agent [
21], plasmid quantity [
22], plasmid desalting [
23], electroporation buffer [
20], and heat treatment [
24]. Some factors have combined effects on electroporation efficiency [
19,
25]. Hence, it is desirable to apply the multifactorial experimental design to investigate the combined effects of the conditions in order to improve electroporation efficiency. Response surface methodology (RSM) is a type of simple, commonly used multifactorial experimental design methodology used for the optimization of a small number of factors. It not only reflects the combined effects of the studied factors, but also allows completion of the optimization through a small number of experiments. To date, RSM design has been widely applied for the optimization of production conditions and media in industrial formulations [
26,
27] and has been rarely used to optimize electroporation conditions.
In the present study, the effects of seven factors on electroporation were investigated, including the growth media, growth phase, electric field, concentration of weakening agent, electroporation buffer, plasmid quantity, as well as heat treatment. In addition, RSM design was applied to investigate the combined effects of wall-weakening agents on the electroporation efficiency. We ultimately developed an efficient electroporation method for B. subtilis ZK that yielded a high efficiency (1.03 × 107 transformants/μg of DNA), which would enable the construction of a large mutant library or the knockout of a gene in wild-type B. subtilis ZK. In order to validate the electroporation efficiency, a gene (rapC) was successfully inactivated using the suicide plasmid pMUTIN4 with the established electroporation method.
3. Experimental Section
3.1. Chemicals
All of chemicals used in this study were purchased from Sangon Biotech (Shanghai, China), Tiangen (Beijing, China) or cdboruike (Chengdu, China).
3.2. Preparation of Desalted Plasmid for Electroporation
Escherichia coli JM110 carrying pHT43 (8057 bp; cat; MoBitech GmbH, Goettingen, Germany) was grown in LB (5 g of yeast extract, 5 g of NaCl, and 10 g of tryptone per litre) with 100 μg·mL
−1 of ampicillin (Amp) at 37 °C, and 180 rpm for 18 h. The pHT43 plasmid was extracted from
E. coli JM110 using the Plasmid Mini Kit (OMEGA Bio-tek, Norcross, GA, USA) and was desalted as described in the literatures [
14,
23]. Plasmid quantification was performed using a spectrophotometer (Nanodrop 2000C, Thermo, Wilmington, MA, USA). After purification, the plasmid was dissolved in ddH
2O and stored at −20 °C.
3.4. The Individual Treatment of Wall-Weakening Agents
A 20% (w/v) glycine solution and a 5% Tween 80 (w/v) solution were individually sterilized at 115 °C for 20 min in advance, and 12.5% dl-threonine solution (w/v) and 50 mg/mL ampicillin were filtered for sterilization. B. subtilis ZK was grown in 40 mL of growth medium. When the optical density at 600 nm (OD600), as monitored using an U-1800 spectrophotometer (Hitachi, Tokyo, Japan) reached 0.5, the glycine, Tween 80, ampicillin, and dl-threonine solutions were added to the growth medium. After shaking at 37 °C and 200 rpm for 1 h, the electro-competent cells were prepared.
3.5. A Combinatorial Treatment of Wall-Weakening Agents
To optimize the concentrations of
X1 (glycine),
X2 (
dl-threonine), and
X3 (Tween 80) on the electroporation efficiency (
Y), a Box-Behnken response surface design (BBD) were performed. A three-variable three-level design was employed, and the values of the variables were selected according to the independent wall-weakening treatment. The coded and un-coded values of the variables are listed in
Table 7. The relationship between the three variables and the electroporation efficiency is expressed by the quadratic polynomial shown in Equation (2):
where
Y is the predicted response (transformants/μg of DNA).
Xi and
Xj are variables. β
0, β
i, β
j, and β
ij are constant, linear, square, and interaction coefficients respectively, and m is the number of variable; The BBD design, analysis of variance (ANOVA) and coefficients of the equation were calculated using the Design expert 8.05b software (Stat-Ease Inc., Minneapolis, MN, USA).
Table 7.
Coded and un-coded values of the variables.
Table 7.
Coded and un-coded values of the variables.
| Variables (%) | Un-Coded | Coded |
|---|
| Gly concentration | 0.55 | −1 |
| 0.65 | 0 |
| 0.75 | 1 |
| dl-Thr concentration | 0.6 | −1 |
| 1 | 0 |
| 1.5 | 1 |
| Tween 80 concentration | 0.02 | −1 |
| 0.06 | 0 |
| 0.1 | 1 |
3.6. Screening of the Electroporation Buffer and Preparation of Electro-Competent Cells
The cells in growth medium were chilled on ice for 10 min and harvested by centrifugation at 10,000× g and 4 °C for 5 min. The cells were purified four times using different buffers, including ice-cold MSG buffer (0.5 M sorbitol, 0.5 M mannitol and 10% glycerol), MKK buffer (0.5 mM MgCl2, 0.25 mM K2HPO4 and 0.25 mM KH2PO4 per litre of water, pH 7.2), SMKK buffer (272 mM sucrose, 0.5 mM MgCl2, 0.5 mM K2HPO4 and 0.5 mM KH2PO4 per litre of water, pH7.2), TSM buffer (0.5 M trehalose, 0.5 M sorbitol, and 0.5 M mannitol per litre of water), and TSMMKK buffer (0.5 M trehalose, 0.5 M sorbitol, 0.5 M mannitol, 0.5 mM MgCl2, 0.5 mM K2HPO4 and 0.5 mM KH2PO4 per litre of water, pH 7.2). After suspension in 1 mL of electroporation buffer, the cells were frozen in liquid nitrogen and stored at −80 °C for later use.
3.7. Electroporation
One aliquot (60 μL) of electro-competent cells was thawed on ice and then mixed with the plasmid. After incubation on ice for 3 min, the mixture was loaded into a prechilled electroporation cuvette (0.1-cm electrode gap) and exposed to a single pulse generated by a Gene Pulser System (Bio-Rad, Hercules, CA, USA). After the electrical pulse, 1 mL of LBMS (0.5 M sorbitol, 10 g of NaCl, 10 g of tryptone, 5 g of yeast extract, 0.5 M mannitol, and 0.38 M sorbitol per litre of water) was added immediately to the mixture. After shaking at 37 °C and 200 rpm for 3 h, the cells were harvested and plated on LB agar plates with 5 μg·mL−1 chloramphenicol (Cm). The transformants were counted following overnight incubation at 37 °C without agitation.
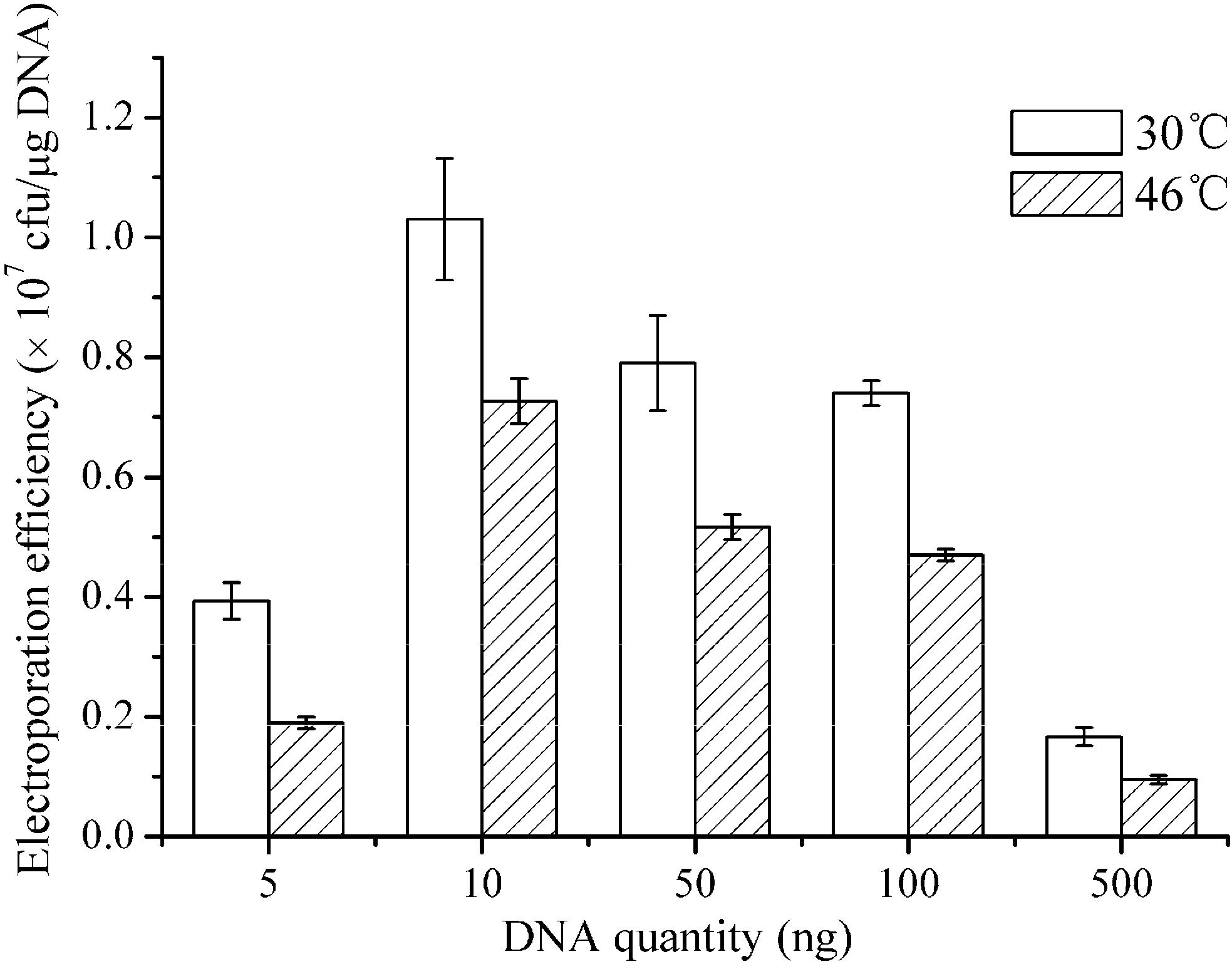
3.8. Treatment of Heat Inactivation
After electroporation, the cells were incubated at 30 or 46 °C for 6 min as previous report [
24].
3.9. Construction of Plasmids and Analysis of Mutants
A 595 bp fragment encompassing middle encoding region of rapC was amplified with primers P-1 (5'-CCCCGGAATTCGGGCTTCTCGATTATTACGTCAACT-3') (with a EcoRI site, underlined) and P-2 (5'-CCCGCGGATCCACGGGATCGTCTGTTTCTTTAGCAT-3') (with a BamHI site, underlined). The fragment was digested with EcoRI and BamHI and then cloned to pMUTIN4 plasmid. Recombinant plasmid was extracted from E. coli and transferred into B. subtilis ZK. The mutants were selected on LB plates containing 0.3 μg·mL−1 erythromycin (Em). The total DNA of mutant was extracted using the bacterial DNA Kit (OMEGA Bio-tek, Norcross, GA, USA). The inactivate mutants were verified by PCR with the specific primers P-3 (5'-ATGAATCATCTTGAAACCGGCAGTC-3') within upstream of the rapC and P-4 (5'-ATCCACGGGATCGTCTGTTTCTTTA-3') within the downstream of the multiple clone site of pMUTIN4.
A 1146 bp full encoding region of rapC was amplified with primers P-5 (5'-ACATCAGCCGTAGGATCCATGAAGAGCGGGTTAATTCCTTCTT-3') (with a BamHI site, underlined) and P-6 (5'-TAGGCGGGCTGCCCCGGGTTAGATTTCAATTTCATACAAACCC-3') (with a SmaI site, underlined). The fragment was digested with BamHI and SmaI and then cloned to pHT01 plasmid. Recombinant plasmid was extracted from E. coli and transferred into rapC mutant. The transformants were selected on LB plates containing 5 μg·mL−1 chloramphenicol (Cm) and 0.3 μg·mL−1 erythromycin (Em).
3.10. Fermentation and Measurement of Iturin A
The ZK strains were activated in LB plates at 37 °C for 36 h. The activated strains were separately inoculated in 5 mL LB mediums (250 mL Erlenmeyer flask) in a shaker at 30 °C with 150 rpm for 22 h. The seed cultures were cultivated in 40 mL LB medium by 10% amount of inoculum at 30 °C, under 150 rpm for 2 days. The extraction and measurement of iturin A as previously reported [
42].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






