
PRRT2 Mutant Leads to Dysfunction of Glutamate Signaling

Abstract
:
1. Introduction
2. Results
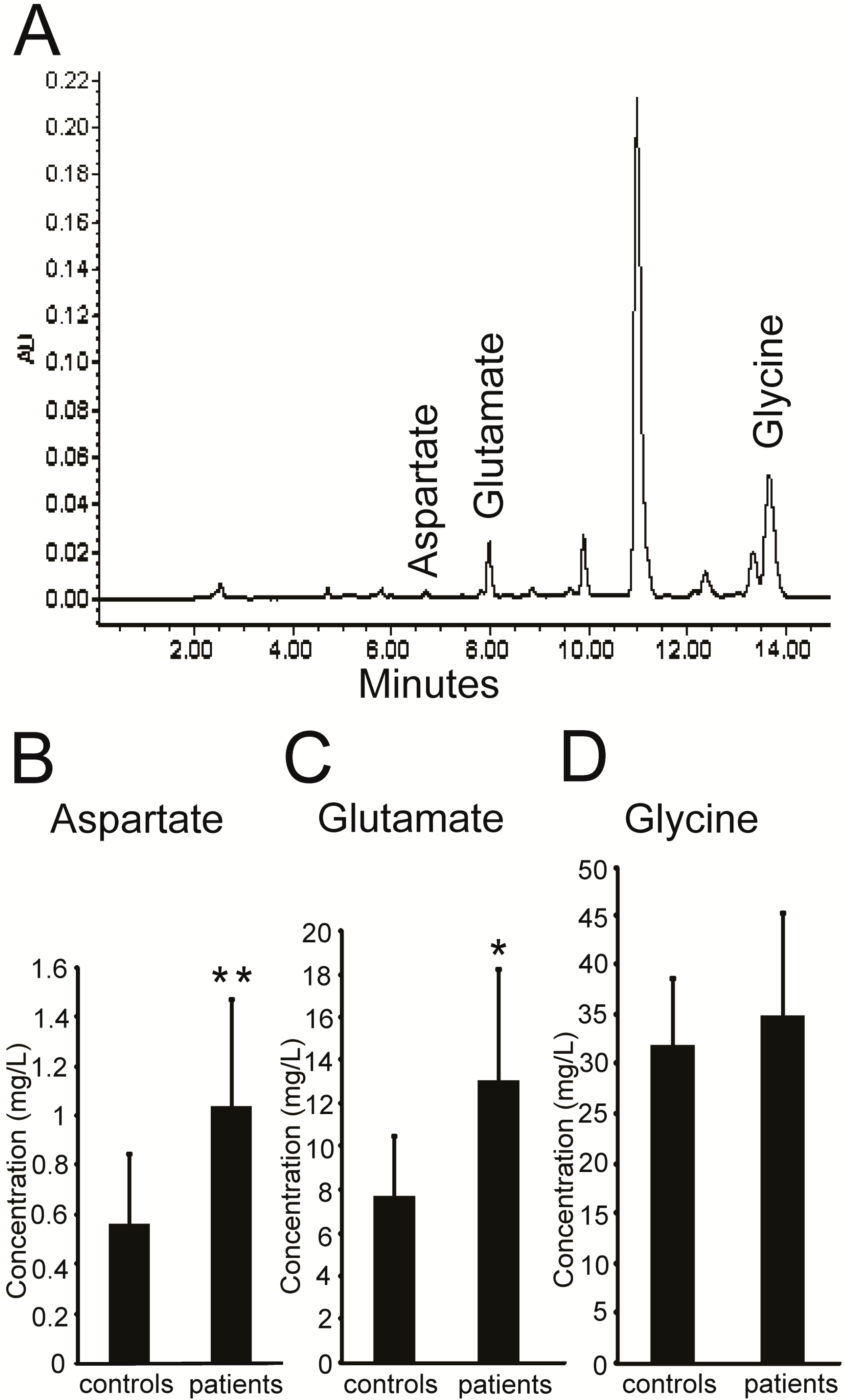
2.1. Higher Levels of Excitatory Amino Acids (EAAs) in the Plasma of PKC Patients


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | PKC Patients (n = 7) | Healthy Controls (n = 12) | |
|---|---|---|---|
| Mean age, year (±SD) | 22.7 ± 2.5 | 27.3 ± 3.0 | |
| Sex | Male (%) | 4 (57.1) | 8 (66.7) |
| Female (%) | 3 (42.9) | 4 (33.3) | |
| Age at onset, year (±SD) | 13.7 ± 2.0 | - | |
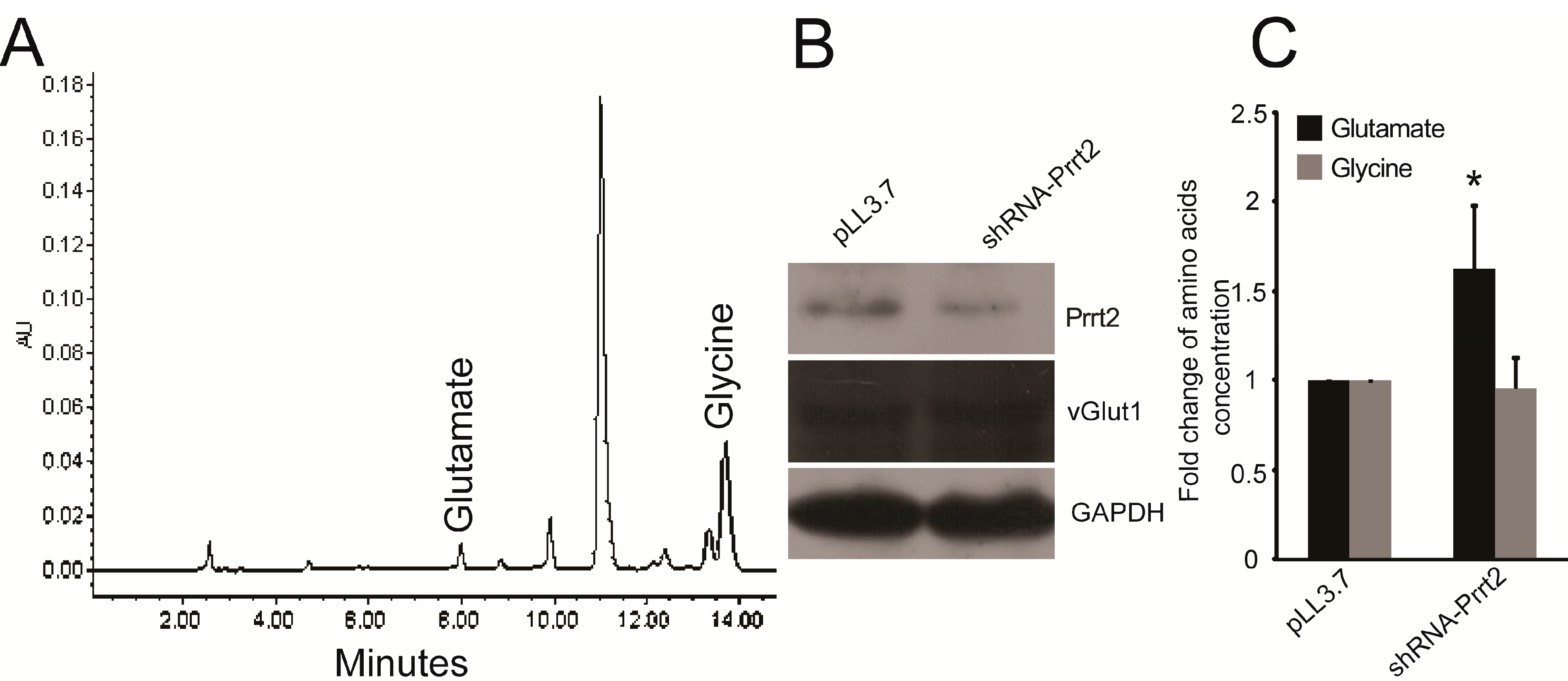
2.2. Knock-out Prrt2 Mice Exhibit Increased Glutamate Level in Neural Cell Culture

2.3. Prrt2 Is Located at the Glutamatergic Neurons

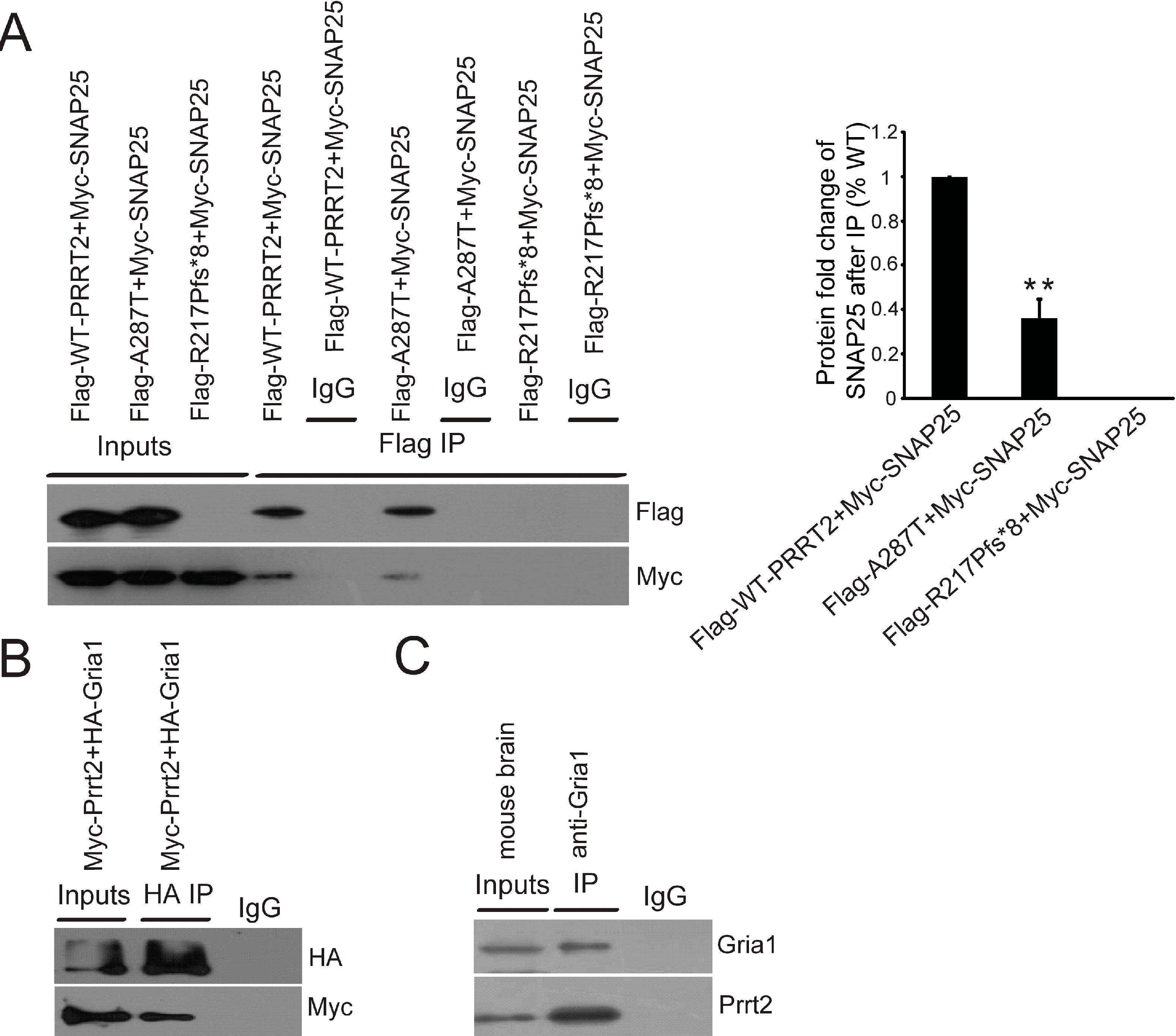
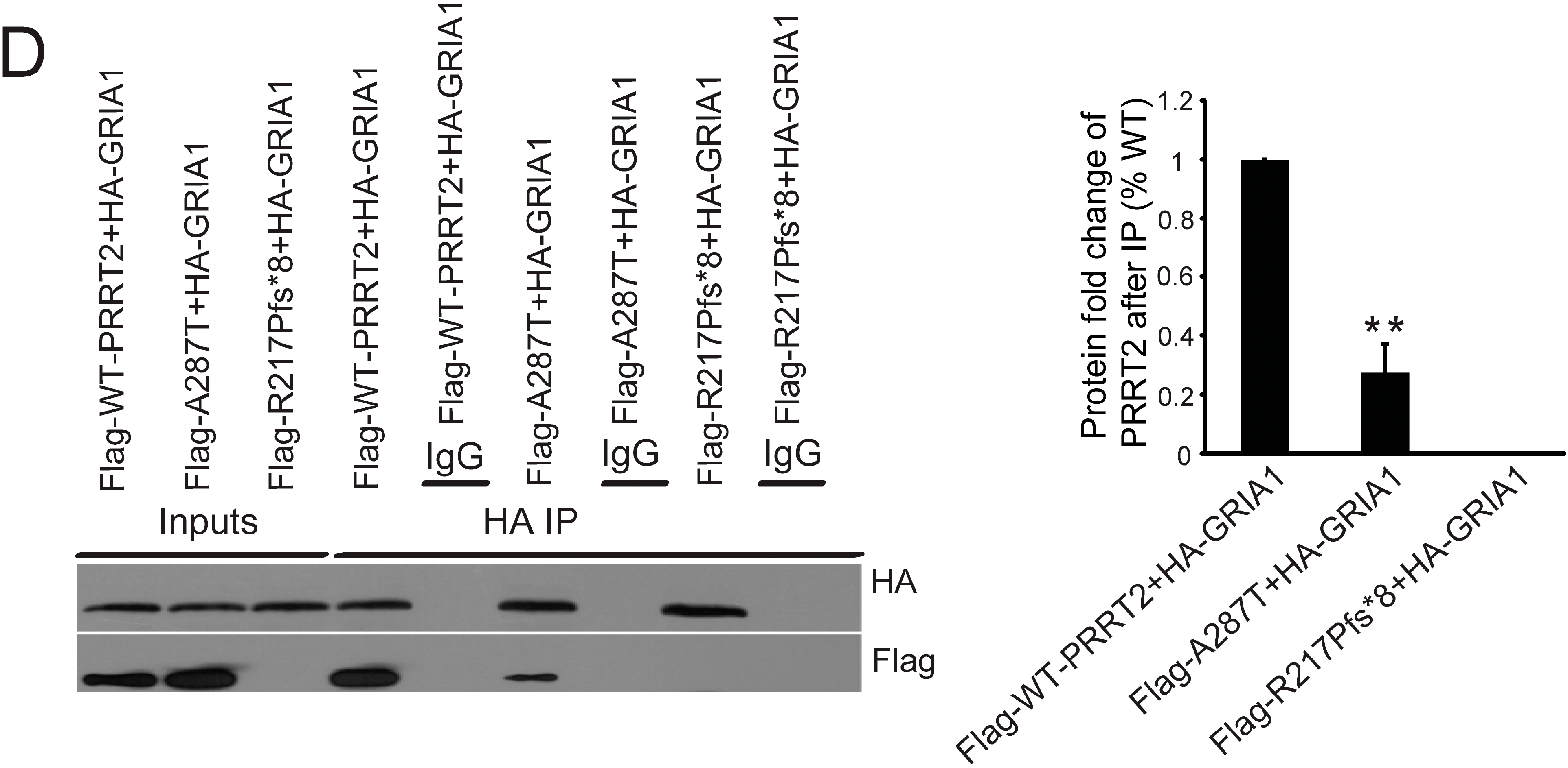
2.4. Interactions between PRRT2 and Its Partners


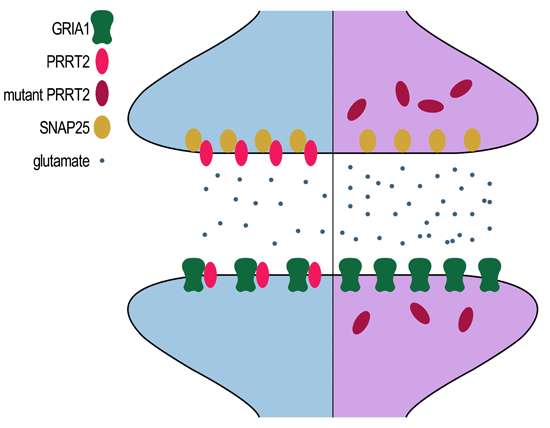
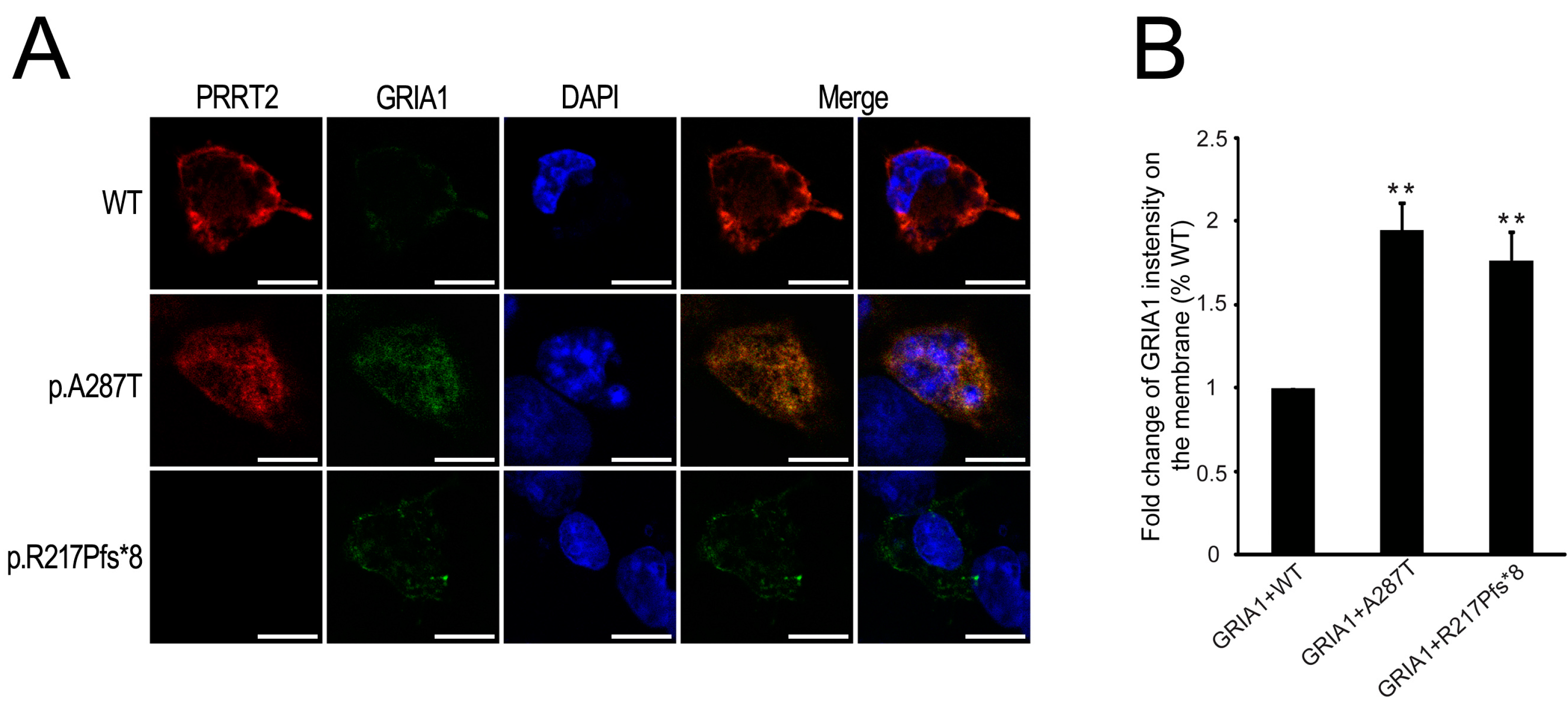
2.5. Mutant PRRT2 Increases Distribution of GRIA1 on the Membrane

3. Discussion

4. Experimental Section
4.1. Chemicals and Antibodies
4.2. Subjects and Plasma Preparation
4.3. Animals
4.4. Aspartate, Glutamate and Glycine Content Measurement
4.5. Cell Culture and Transfection
4.6. Plasmid Constructions
4.7. Recombinant Lentivirus Infection
4.8. Co-Immunoprecipitation
4.9. Western Blotting
4.10. Immunofluorescence Experiments
4.11. Live-Labeling
4.12. Statistical Analysis
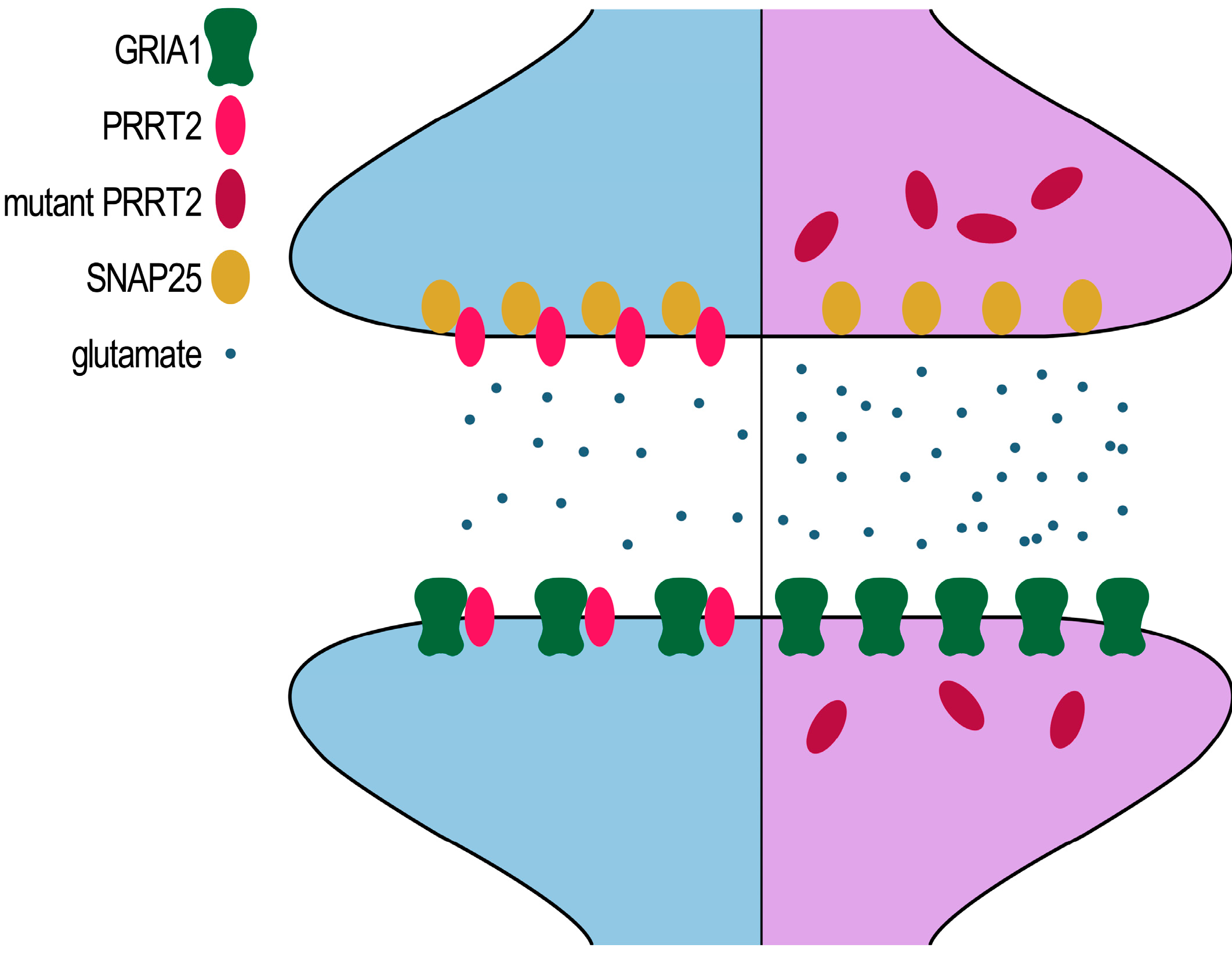
5. Conclusions
Supplementary Information
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kertesz, A. Paroxysmal kinesigenic choreoathetosis. An entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology 1967, 17, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.K.; Hallett, M.; Gwinn-Hardy, K.; Sorensen, B.; Considine, E.; Tucker, S.; Lynch, D.R.; Mathews, K.D.; Swoboda, K.J.; Harris, J.; et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: New diagnostic criteria. Neurology 2004, 63, 2280–2287. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, X.; Wang, X.; Sun, W.; Feng, B.; Du, T.; Sun, B.; Niu, F.; Wei, H.; Wu, X.; et al. Targeted genomic sequencing identifies PRRT2 mutations as a cause of paroxysmal kinesigenic choreoathetosis. J. Med. Genet. 2012, 49, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Lin, Y.; Xiong, Z.Q.; Wei, W.; Ni, W.; Tan, G.H.; Guo, S.L.; He, J.; Chen, Y.F.; Zhang, Q.J.; et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat. Genet. 2011, 43, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Cao, L.; Li, X.H.; Hu, Z.M.; Li, J.D.; Zhang, J.G.; Liang, Y.; San, A.; Li, N.; Chen, S.Q.; et al. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain 2011, 134, 3493–3501. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Huang, Y.; Bruneau, N.; Roll, P.; Roberson, E.D.; Hermann, M.; Quinn, E.; Maas, J.; Edwards, R.; Ashizawa, T.; et al. Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell. Rep. 2012, 1, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Grinton, B.E.; Kivity, S.; Afawi, Z.; Zuberi, S.M.; Hughes, J.N.; Pridmore, C.; Hodgson, B.L.; Iona, X.; Sadleir, L.G.; et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am. J. Hum. Genet. 2012, 90, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xia, Z.; Liu, Y. SNAP-25 functional domains in SNARE core complex assembly and glutamate release of cerebellar granule cells. J. Biol. Chem. 2000, 275, 29482–29487. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Pozzi, D.; Pravettoni, E.; Inverardi, F.; Schenk, U.; Coco, S.; Proux-Gillardeaux, V.; Galli, T.; Rossetto, O.; Frassoni, C.; et al. SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron 2004, 41, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Garbelli, R.; Inverardi, F.; Medici, V.; Amadeo, A.; Verderio, C.; Matteoli, M.; Frassoni, C. Heterogeneous expression of SNAP-25 in rat and human brain. J. Comp. Neurol. 2008, 506, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; Vanni, V.; Cesa, R.; Grasselli, G.; Puglisi, F.; Cesare, P.; Strata, P. Distribution of the SNAP25 and SNAP23 synaptosomal-associated protein isoforms in rat cerebellar cortex. Neuroscience 2009, 164, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Costantin, L.; Bozzi, Y.; Richichi, C.; Viegi, A.; Antonucci, F.; Funicello, M.; Gobbi, M.; Mennini, T.; Rossetto, O.; Montecucco, C.; et al. Antiepileptic effects of botulinum neurotoxin E. J. Neurosci. 2005, 25, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Haglid, K.G.; Wang, S.; Qiner, Y.; Hamberger, A. Excitotoxicity. Experimental correlates to human epilepsy. Mol. Neurobiol. 1994, 9, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Alam, Z.; Coombes, N.; Waring, R.H.; Williams, A.C.; Steventon, G.B. Plasma levels of neuroexcitatory amino acids in patients with migraine or tension headache. J. Neurol. Sci. 1998, 156, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Shimmura, C.; Suda, S.; Tsuchiya, K.J.; Hashimoto, K.; Ohno, K.; Matsuzaki, H.; Iwata, K.; Matsumoto, K.; Wakuda, T.; Kameno, Y.; et al. Alteration of plasma glutamate and glutamine levels in children with high-functioning autism. PLoS ONE 2011, 6, e25340. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, J.; Harmel, N.; Brechet, A.; Zolles, G.; Berkefeld, H.; Muller, C.S.; Bildl, W.; Baehrens, D.; Huber, B.; Kulik, A.; et al. High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron 2012, 74, 621–633. [Google Scholar] [CrossRef] [PubMed]
- McGale, E.H.; Pye, I.F.; Stonier, C.; Hutchinson, E.C.; Aber, G.M. Studies of the inter-relationship between cerebrospinal fluid and plasma amino acid concentrations in normal individuals. J. Neurochem. 1977, 29, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Alfredsson, G.; Wiesel, F.A.; Tylec, A. Relationships between glutamate and monoamine metabolites in cerebrospinal fluid and serum in healthy volunteers. Biol. Psychiatry 1988, 23, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Gerachshenko, T.; Blackmer, T.; Yoon, E.J.; Bartleson, C.; Hamm, H.E.; Alford, S. Gbetagamma acts at the C terminus of SNAP-25 to mediate presynaptic inhibition. Nat. Neurosci. 2005, 8, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Abdullah, J.M. The role of GluA1 in central nervous system disorders. Rev. Neurosci. 2013, 24, 499–505. [Google Scholar] [PubMed]
- Almen, M.S.; Bringeland, N.; Fredriksson, R.; Schioth, H.B. The dispanins: A novel gene family of ancient origin that contains 14 human members. PLoS ONE 2012, 7, e31961. [Google Scholar] [CrossRef]
- Kalashnikova, E.; Lorca, R.A.; Kaur, I.; Barisone, G.A.; Li, B.; Ishimaru, T.; Trimmer, J.S.; Mohapatra, D.P.; Diaz, E. SynDIG1: An activity-regulated, AMPA-receptor-interacting transmembrane protein that regulates excitatory synapse development. Neuron 2010, 65, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Dibbens, L.M. Role of PRRT2 in common paroxysmal neurological disorders: A gene with remarkable pleiotropy. J. Med. Genet. 2013, 50, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, J.; Deng, X.; Xiao, J.; Yuan, L.; Yang, Y.; Guan, L.; Song, Z.; Yang, Z.; Deng, H. Identification of a premature termination mutation in the Proline-Rich Transmembrane Protein 2 Gene in a Chinese Family with Febrile Seizures. Mol. Neurobiol. 2014, in press. [Google Scholar]
- He, Z.W.; Qu, J.; Zhang, Y.; Mao, C.X.; Wang, Z.B.; Mao, X.Y.; Deng, Z.Y.; Zhou, B.T.; Yin, J.Y.; Long, H.Y.; et al. PRRT2 Mutations Are related to febrile Seizures in Epileptic Patients. Int. J. Mol. Sci. 2014, 15, 23408–23417. [Google Scholar] [CrossRef] [PubMed]
- Meneret, A.; Gaudebout, C.; Riant, F.; Vidailhet, M.; Depienne, C.; Roze, E. PRRT2 mutations and paroxysmal disorders. Eur. J. Neurol. 2013, 20, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Delcourt, M.; Riant, F.; Mancini, J.; Milh, M.; Navarro, V.; Roze, E.; Humbertclaude, V.; Korff, C.; Portes, V.D.; Szepetowski, P.; et al. Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. J. Neurol. Neurosurg. Psychiatry 2015, in press. [Google Scholar]
- Eid, T.; Thomas, M.J.; Spencer, D.D.; Runden-Pran, E.; Lai, J.C.; Malthankar, G.V.; Kim, J.H.; Danbolt, N.C.; Ottersen, O.P.; Lanerolle, N.C. Loss of glutamine synthetase in the human epileptogenic hippocampus: Possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 2004, 363, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Cavus, I.; Pan, J.W.; Hetherington, H.P.; Abi-Saab, W.; Zaveri, H.P.; Vives, K.P.; Krystal, J.H.; Spencer, S.S.; Spencer, D.D. Decreased hippocampal volume on MRI is associated with increased extracellular glutamate in epilepsy patients. Epilepsia 2008, 49, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Sirvanci, S.; Meshul, C.K.; Onat, F.; San, T. Immunocytochemical analysis of glutamate and GABA in hippocampus of genetic absence epilepsy rats (GAERS). Brain Res. 2003, 988, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Podell, M.; Hadjiconstantinou, M. Cerebrospinal fluid gamma-aminobutyric acid and glutamate values in dogs with epilepsy. Am. J. Vet. Res. 1997, 58, 451–456. [Google Scholar] [PubMed]
- Creevy, K.E.; Gagnepain, J.F.; Platt, S.R.; Edwards, G.L.; Kent, M. Comparison of concentrations of γ-aminobutyric acid and glutamate in cerebrospinal fluid of dogs with idiopathic epilepsy with and without seizure-related magnetic resonance imaging hyperintense areas in the limbic system. Am. J. Vet. Res. 2013, 74, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S. The role of glutamate in epilepsy and other CNS disorders. Neurology 1994, 44, S14–S23. [Google Scholar] [PubMed]
- Paraskevas, G.P.; Triantafyllou, N.I.; Kapaki, E.; Limpitaki, G.; Petropoulou, O.; Vassilopoulos, D. Add-on lamotrigine treatment and plasma glutamate levels in epilepsy: Relation to treatment response. Epilepsy Res. 2006, 70, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.D.; Odink, J.; Bos, K.D.; Malessy, M.J.; Bruyn, G.W. Neuroexcitatory plasma amino acids are elevated in migraine. Neurology 1990, 40, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Cananzi, A.R.; D’Andrea, G.; Perini, F.; Zamberlan, F.; Welch, K.M. Platelet and plasma levels of glutamate and glutamine in migraine with and without aura. Cephalalgia 1995, 15, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Gallai, V.; Alberti, A.; Gallai, B.; Coppola, F.; Floridi, A.; Sarchielli, P. Glutamate and nitric oxide pathway in chronic daily headache: Evidence from cerebrospinal fluid. Cephalalgia 2003, 23, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Peres, M.F.; Zukerman, E.; Senne Soares, C.A.; Alonso, E.O.; Santos, B.F.; Faulhaber, M.H. Cerebrospinal fluid glutamate levels in chronic migraine. Cephalalgia 2004, 24, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Vieira, D.S.; Naffah-Mazzacoratti Mda, G.; Zukerman, E.; Senne Soares, C.A.; Cavalheiro, E.A.; Peres, M.F. Glutamate levels in cerebrospinal fluid and triptans overuse in chronic migraine. Headache 2007, 47, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Spaccapelo, L.; Pinetti, D.; Tacchi, R.; Bertolini, A. Effective prophylactic treatments of migraine lower plasma glutamate levels. Cephalalgia 2009, 29, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Stover, J.F.; Kempski, O.S. Anesthesia increases circulating glutamate in neurosurgical patients. Acta Neurochir. (Wien.) 2005, 147, 847–853. [Google Scholar] [CrossRef]
- Friedman, A.; Kaufer, D.; Shemer, J.; Hendler, I.; Soreq, H.; Tur-Kaspa, I. Pyridostigmine brain penetration under stress enhances neuronal excitability and induces early immediate transcriptional response. Nat. Med. 1996, 2, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Mayhan, W.G.; Didion, S.P. Glutamate-induced disruption of the blood-brain barrier in rats. Role of nitric oxide. Stroke 1996, 27, 965–969, discussion 970. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Nakayama, J.; Xu, Y.; Fan, X.; Karouani, M.; Shen, Y.; Pothos, E.N.; Hess, E.J.; Fu, Y.H.; Edwards, R.H.; et al. Dopamine dysregulation in a mouse model of paroxysmal nonkinesigenic dyskinesia. J. Clin. Investig. 2012, 122, 507–518. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Kang, K.S.; Kotzaeridou, U.; Kohlhase, J.; Klein, C.; Assmann, B.E. Child Neurology: PRRT2-associated movement disorders and differential diagnoses. Neurology 2014, 83, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.P.; Battenberg, E.; Wilson, M.C. SNAP-25 and synaptotagmin involvement in the final Ca2+-dependent triggering of neurotransmitter exocytosis. Proc. Natl. Acad. Sci. USA 1996, 93, 10471–10476. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Cerri, C.; Maya Vetencourt, J.F.; Caleo, M. Acute neuroprotection by the synaptic blocker botulinum neurotoxin E in a rat model of focal cerebral ischaemia. Neuroscience 2010, 169, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Dolly, J.O.; O’Connell, M.A. Neurotherapeutics to inhibit exocytosis from sensory neurons for the control of chronic pain. Curr. Opin. Pharmacol. 2012, 12, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Kennard, J.T.; Barmanray, R.; Sampurno, S.; Ozturk, E.; Reid, C.A.; Paradiso, L.; D’Abaco, G.M.; Kaye, A.H.; Foote, S.J.; O’Brien, T.J.; et al. Stargazin and AMPA receptor membrane expression is increased in the somatosensory cortex of Genetic Absence Epilepsy Rats from Strasbourg. Neurobiol. Dis. 2011, 42, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, K.; Joshi, S.; Kozhemyakin, M.; Todorovic, M.S.; Kowalski, S.; Balint, C.; Kapur, J. Receptor trafficking hypothesis revisited: Plasticity of AMPA receptors during established status epilepticus. Epilepsia 2013, 54, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Formicola, D.; Aloia, A.; Sampaolo, S.; Farina, O.; Diodato, D.; Griffiths, L.R.; Gianfrancesco, F.; di Iorio, G.; Esposito, T. Common variants in the regulative regions of GRIA1 and GRIA3 receptor genes are associated with migraine susceptibility. BMC Med. Genet. 2010, 11. [Google Scholar] [CrossRef]
- Faught, E. BGG492 (selurampanel), an AMPA/kainate receptor antagonist drug for epilepsy. Expert Opin. Investig. Drugs 2014, 23, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Mancilla, B.; Brand, R.; Jurgens, T.P.; Gobel, H.; Sommer, C.; Straube, A.; Evers, S.; Sommer, M.; Campos, V.; Kalkman, H.O.; et al. Randomized, multicenter trial to assess the efficacy, safety and tolerability of a single dose of a novel AMPA receptor antagonist BGG492 for the treatment of acute migraine attacks. Cephalalgia 2014, 34, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Li, H.F.; Chen, W.J.; Ni, W.; Wang, K.Y.; Liu, G.L.; Wang, N.; Xiong, Z.Q.; Xu, J.; Wu, Z.Y. PRRT2 mutation correlated with phenotype of paroxysmal kinesigenic dyskinesia and drug response. Neurology 2013, 80, 1534–1535. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Wu, Z.; Wang, J.; Wang, X.; Jia, S.; Bazer, F.W.; Wu, G. Analysis of polyamines in biological samples by HPLC involving pre-column derivatization with O-phthalaldehyde and N-acetyl-l-cysteine. Amino Acids 2014, 46, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, Y.; Wu, J.; Lv, G.; Zhou, L.; Jia, J. Effect of Buyang Huanwu decoction on amino acid content in cerebrospinal fluid of rats during ischemic/reperfusion injury. J. Pharm. Biomed. Anal. 2013, 86, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, L.; You, Y.; Gong, Y.; Yin, B.; Yuan, J.; Peng, X. Downstream of tyrosine kinase/docking protein 6, as a novel substrate of tropomyosin-related kinase C receptor, is involved in neurotrophin 3-mediated neurite outgrowth in mouse cortex neurons. BMC Biol. 2010, 8. [Google Scholar] [CrossRef]
- Rubinson, D.A.; Dillon, C.P.; Kwiatkowski, A.V.; Sievers, C.; Yang, L.; Kopinja, J.; Rooney, D.L.; Zhang, M.; Ihrig, M.M.; McManus, M.T.; et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 2003, 33, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, X.; Liu, K.; Zhang, X.; Yang, F.; Zhang, H.; He, Y.; Zhu, T.; Li, F.; Shi, W.; et al. NOX2 deficiency ameliorates cerebral injury through reduction of complexin II-mediated glutamate excitotoxicity in experimental stroke. Free Radic. Biol. Med. 2013, 65, 942–951. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Niu, F.; Zhu, X.; Wu, X.; Shen, N.; Peng, X.; Liu, Y. PRRT2 Mutant Leads to Dysfunction of Glutamate Signaling. Int. J. Mol. Sci. 2015, 16, 9134-9151. https://doi.org/10.3390/ijms16059134
Li M, Niu F, Zhu X, Wu X, Shen N, Peng X, Liu Y. PRRT2 Mutant Leads to Dysfunction of Glutamate Signaling. International Journal of Molecular Sciences. 2015; 16(5):9134-9151. https://doi.org/10.3390/ijms16059134
Chicago/Turabian StyleLi, Ming, Fenghe Niu, Xilin Zhu, Xiaopan Wu, Ning Shen, Xiaozhong Peng, and Ying Liu. 2015. "PRRT2 Mutant Leads to Dysfunction of Glutamate Signaling" International Journal of Molecular Sciences 16, no. 5: 9134-9151. https://doi.org/10.3390/ijms16059134
APA StyleLi, M., Niu, F., Zhu, X., Wu, X., Shen, N., Peng, X., & Liu, Y. (2015). PRRT2 Mutant Leads to Dysfunction of Glutamate Signaling. International Journal of Molecular Sciences, 16(5), 9134-9151. https://doi.org/10.3390/ijms16059134