
Pathogenesis of Brain Edema and Investigation into Anti-Edema Drugs

Abstract
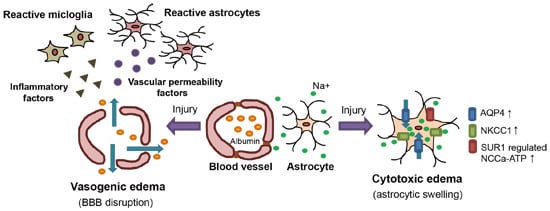
:
1. Introduction
2. Classification of Brain Edema

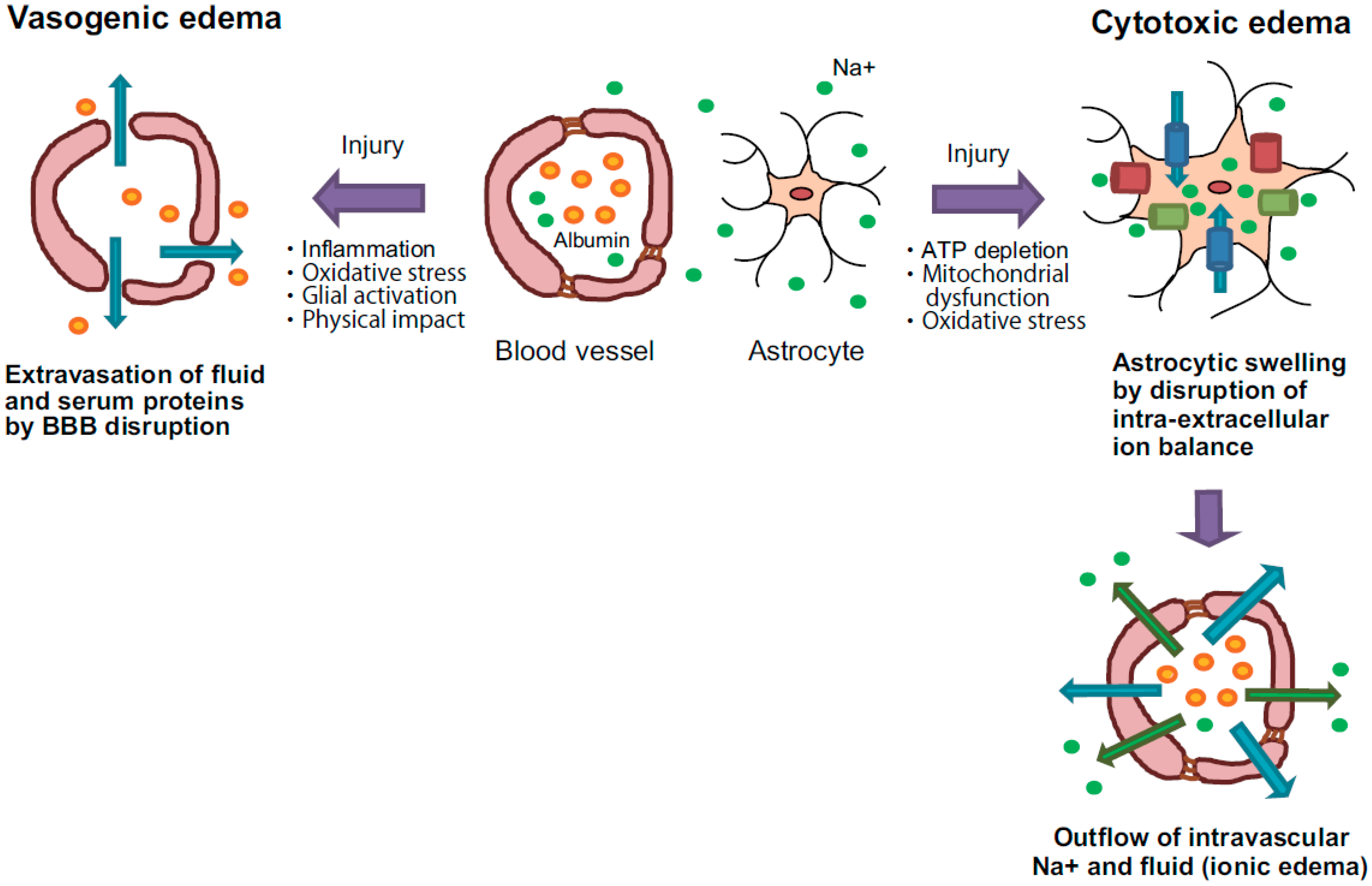
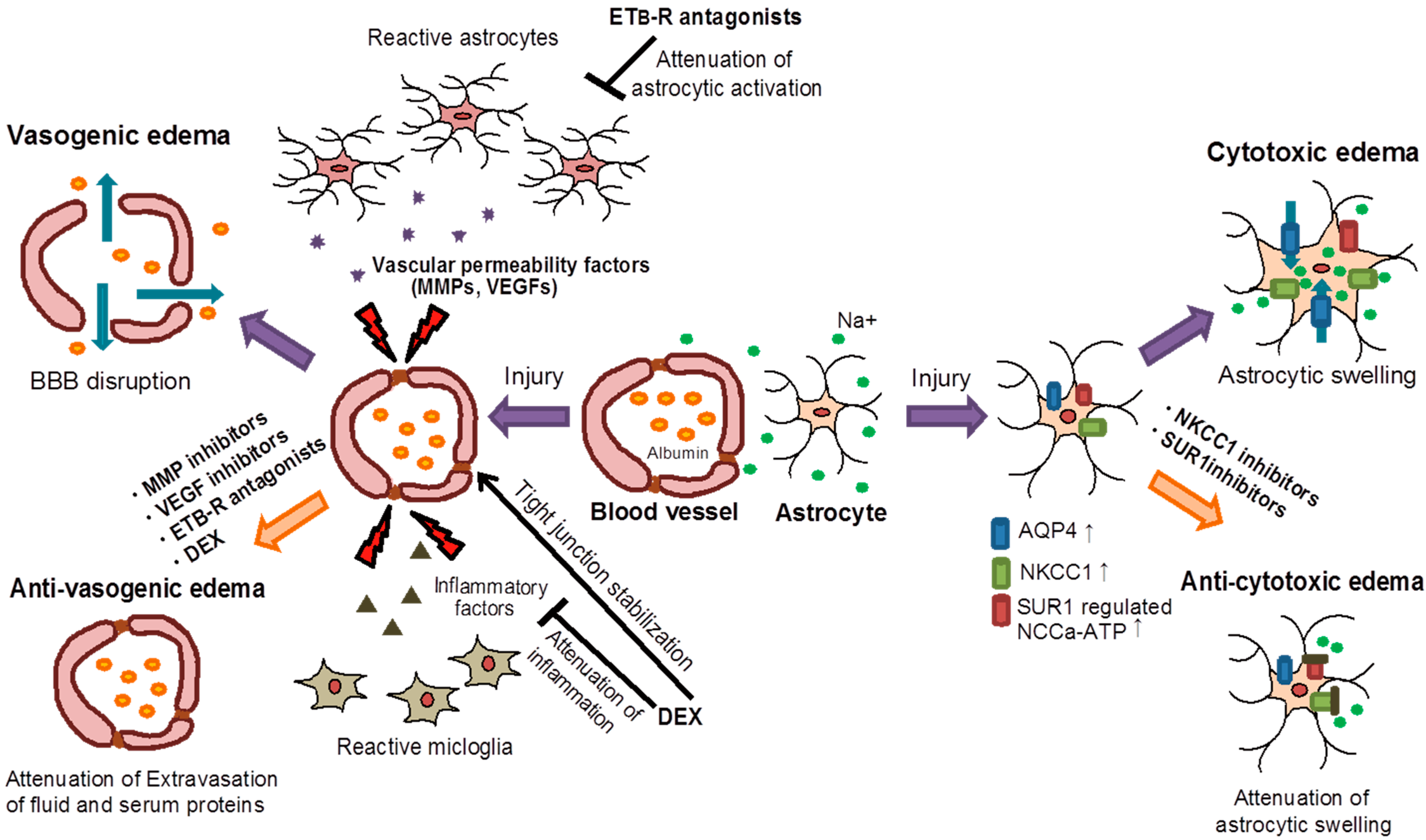
2.1. Vasogenic Edema
2.2. Cytotoxic Edema
3. Experimental Models of Brain Edema in Animals
3.1. The Cold Injury Model

3.2. The Fluid Percussion Injury (FPI) Model
3.3. The Cerebral Hemorrhage Model
3.4. The Water Intoxication Model
3.5. The Liver Failure Model
4. Methods for Evaluating Brain Edema
4.1. Wet-Dry Weight Method
4.2. The Gravimetric Method
4.3. Magnetic Resonance Imaging (MRI)
5. Key Molecules of Brain Edema Formation: Possible Targets of Anti-Edema Drugs
5.1. Vascular Endothelial Growth Factors (VEGFs)

5.2. Matrixmetalloproteinases (MMPs)
5.3. Aquaporins (AQPs)
5.4. Na+–K+–Cl−–Co-Transporter 1 (NKCC1)
5.5. Sulfonylurea Receptor 1 (SUR1)-Regulated Nonselective Cation Channels (NCCa-ATP)
5.6. Endothelin ETB Receptor (ETB-R)
5.7. Glucocorticoid Receptor (GR)
6. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Candidates of Anti-Edema Drugs | |
|---|---|
| Anti-vasogenic edema drugs | Anti-cytotoxic edema drugs |
| MMP inhibitors | KNCC1inhibitors (bumetanide) |
| VEGF inhibitors VEGF antibodies | SUR1-regulated NCCa-ATP inhibitors (glibenclamide) |
| ETB-R antagonists | – |
| Glucocorticoids (dexamethasone) | – |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nag, S.; Manias, J.L.; Stewart, D.J. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. 2009, 118, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Bosoi, C.R.; Rose, C.F. Brain edema in acute liver failure and chronic liver disease: Similarities and differences. Neurochem. Int. 2013, 62, 446–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raboel, P.H.; Bartek, J., Jr.; Andresen, M.; Bellander, B.M.; Romner, B. Intracranial pressure monitoring: Invasive versus non-invasive method—A review. Crit. Care Res. Pract. 2012, 2012, 950393. [Google Scholar] [PubMed]
- Unterberg, A.W.; Stover, J.; Kress, B.; Kiening, K.L. Edema and brain trauma. Neuroscience 2004, 4, 1021–1029. [Google Scholar]
- Simard, J.M.; Kent, T.A.; Chen, M.; Tarasov, K.V.; Gerzanich, V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007, 6, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Kuramatsu, J.B.; Huttner, H.B.; Schwab, S. Advances in the management of intracerebral hemorrhage. J. Neural Transm. 2013, 120, S35–S41. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Sharifi, Y.; Jover-Cobos, M.; Jalan, R. The brain in acute on chronic liver failure. Metab. Brain Dis. 2014, 4, 965–973. [Google Scholar] [CrossRef]
- Rabinstein, A.A. Treatment of cerebral edema. Neurologist 2006, 12, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Raslan, A.; Bhardwaj, A. Medical management of cerebral edema. Neurosurg. Focus 2007, 5, E12. [Google Scholar]
- Walcott, B.P.; Kahle, K.T.; Simard, J.M. Novel treatment targets for cerebral edema. Neurotherapeutics 2012, 9, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Thrane, A.S.; Rangroo Thrane, V.; Nedergaard, M. Drowning stars: Reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014, 11, 620–628. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Noell, S.; Mack, A.; Wolburg-Buchholz, K.; Fallier-Becker, P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009, 335, 75–96. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 1, 1–13. [Google Scholar] [CrossRef]
- Paschen, W. Glutamate excitotoxicity in transient global cerebral ischemia. Acta Neurobiol. Exp. (Wars) 1996, 1, 313–322. [Google Scholar]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 4, 699–719. [Google Scholar] [CrossRef]
- Walker, K.R.; Tesco, G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci. 2013, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 2009, 4, 842–857. [Google Scholar] [CrossRef]
- Nedergaard, M.; Dirnagl, U. Role of glial cells in cerebral ischemia. Glia 2005, 4, 281–286. [Google Scholar] [CrossRef]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 2007, 4, 1345–1353. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 6, 977–982. [Google Scholar]
- Kahle, K.T.; Simard, J.M.; Staley, K.J.; Nahed, B.V.; Jones, P.S.; Sun, D. Molecular mechanisms of ischemic cerebral edema: Role of electroneutral ion transport. Physiology (Bethesda) 2009, 24, 257–265. [Google Scholar] [CrossRef]
- Rabinstein, A.A. Treatment of brain edema in acute liver failure. Curr. Treat. Opt. Neurol. 2010, 2, 129–141. [Google Scholar] [CrossRef]
- Liang, D.; Bhatta, S.; Gerzanich, V.; Simard, J.M. Cytotoxic edema: Mechanisms of pathological cell swelling. Neurosurg. Focus 2007, 5, E2. [Google Scholar]
- Oury, T.D.; Piantadosi, C.A.; Crapo, J.D. Cold-induced brain edema in mice. Involvement of extracellular superoxide dismutase and nitric oxide. J. Biol. Chem. 1993, 268, 15394–15398. [Google Scholar] [PubMed]
- Nag, S. Cold-injury of the cerebral cortex: Immunolocalization of cellular proteins and blood-brain barrier permeability studies. J. Neuropathol. Exp. Neurol. 1996, 55, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kondo, T.; Yang, G.; Chen, S.F.; Morita-Fujimura, Y.; Chan, P.H. Cold injury in mice: A model to study mechanisms of brain edema and neuronal apoptosis. Prog. Neurobiol. 1999, 57, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Raslan, F.; Schwarz, T.; Meuth, S.G.; Austinat, M.; Bader, M.; Renné, T.; Roosen, K.; Stoll, G.; Sirén, A.L.; Kleinschnitz, C. Inhibition of bradykinin receptor B1 protects mice from focal brain injury by reducing blood-brain barrier leakage and inflammation. J. Cereb. Blood Flow Metab. 2010, 8, 1477–1486. [Google Scholar] [CrossRef]
- Pifarré, P.; Prado, J.; Giralt, M.; Molinero, A.; Hidalgo, J.; Garcia, A. Cyclic GMP phosphodiesterase inhibition alters the glial inflammatory response, reduces oxidative stress and cell death and increases angiogenesis following focal brain injury. J. Neurochem. 2010, 3, 807–817. [Google Scholar] [CrossRef]
- Donkin, J.J.; Vink, R. Mechanisms of cerebral edema in traumatic brain injury: Therapeutic developments. Curr. Opin. Neurol. 2010, 3, 293–299. [Google Scholar] [CrossRef]
- Xu, M.; Su, W.; Xu, Q.P. Aquaporin-4 and traumatic brain edema. Chin. J. Traumatol. 2010, 2, 103–110. [Google Scholar]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 4, 492–516. [Google Scholar] [CrossRef]
- Nag, S.; Kapadia, A.; Stewart, D.J. Review: Molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol. Appl. Neurobiol. 2011, 1, 3–23. [Google Scholar] [CrossRef]
- Albert-Weissenberger, C.; Sirén, A.L. Experimental traumatic brain injury. Exp. Transl. Stroke Med. 2010, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Lifshitz, J.; Marklund, N.; Grady, M.S.; Graham, D.I.; Hovda, D.A.; McIntosh, T.K. Lateral fluid percussion brain injury: A 15-year review and evaluation. J. Neurotrauma 2005, 1, 42–75. [Google Scholar] [CrossRef]
- Alder, J.; Fujioka, W.; Lifshitz, J.; Crockett, D.P.; Thakker-Varia, S. Lateral fluid percussion: Model of traumatic brain injury in mice. J. Vis. Exp. 2011, 54. [Google Scholar] [CrossRef]
- Maegele, M.; Stuermer, E.K.; Hoeffgen, A.; Uhlenkueken, U.; Mautes, A.; Schaefer, N.; Lippert-Gruener, M.; Schaefer, U.; Hoehn, M. Multimodal MR imaging of acute and subacute experimental traumatic brain injury: Time course and correlation with cerebral energy metabolites. Acta Radiol. Short Rep. 2015, 4. [Google Scholar] [CrossRef]
- Schmidt, R.H.; Grady, M.S. Regional patterns of blood-brain barrier breakdown following central and lateral fluid percussion injury in rodents. J. Neurotrauma 1993, 4, 415–430. [Google Scholar] [CrossRef]
- Fukuda, K.; Tanno, H.; Okimura, Y.; Nakamura, M.; Yamaura, A. The blood-brain barrier disruption to circulating proteins in the early period after fluid percussion brain injury in rats. J. Neurotrauma 1995, 3, 315–324. [Google Scholar] [CrossRef]
- Louin, G.; Marchand-Verrecchia, C.; Palmier, B.; Plotkine, M.; Jafarian-Tehrani, M. Selective inhibition of inducible nitric oxide synthase reduces neurological deficit but not cerebral edema following traumatic brain injury. Neuropharmacology 2006, 2, 182–190. [Google Scholar] [CrossRef]
- Simard, J.M.; Kilbourne, M.; Tsymbalyuk, O.; Tosun, C.; Caridi, J.; Ivanova, S.; Keledjian, K.; Bochicchio, G.; Gerzanich, V. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J. Neurotrauma 2009, 12, 2257–2267. [Google Scholar] [CrossRef]
- Lin, Y.; Pan, Y.; Wang, M.; Huang, X.; Yin, Y.; Wang, Y.; Jia, F.; Xiong, W.; Zhang, N.; Jiang, J.Y. Blood-brain barrier permeability is positively correlated with cerebral microvascular perfusion in the early fluid percussion-injured brain of the rat. Lab. Investig. 2012, 11, 1623–1634. [Google Scholar] [CrossRef]
- Knoblach, S.M.; Faden, A.I. Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimental traumatic brain injury. Exp. Neurol. 1998, 1, 143–151. [Google Scholar] [CrossRef]
- Farias, S.; Frey, L.C.; Murphy, R.C.; Heidenreich, K.A. Injury-related production of cysteinyl leukotrienes contributes to brain damage following experimental traumatic brain injury. J. Neurotrauma 2009, 11, 1977–1986. [Google Scholar] [CrossRef]
- Lotocki, G.; de Rivero Vaccari, J.P.; Perez, E.R.; Sanchez-Molano, J.; Furones-Alonso, O.; Bramlett, H.M.; Dietrich, W.D. Alterations in blood-brain barrier permeability to large and small molecules and leukocyte accumulation after traumatic brain injury: Effects of post-traumatic hypothermia. J. Neurotrauma 2009, 7, 1123–1134. [Google Scholar] [CrossRef]
- Van Putten, H.P.; Bouwhuis, M.G.; Muizelaar, J.P.; Lyeth, B.G.; Berman, R.F. Diffusion-weighted imaging of edema following traumatic brain injury in rats: Effects of secondary hypoxia. J. Neurotrauma 2005, 8, 857–872. [Google Scholar] [CrossRef]
- Claassen, J.; Carhuapoma, J.R.; Kreiter, K.T.; Du, E.Y.; Connolly, E.S.; Mayer, S.A. Global cerebral edema after subarachnoid hemorrhage: Frequency, predictors, and impact on outcome. Stroke 2002, 5, 1225–1232. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Mendelow, A.D.; Hanley, D.F. Intracerebral haemorrhage. Lancet 2009, 373, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.I.; Tuhrim, S.; Broderick, J.P.; Batjer, H.H.; Hondo, H.; Hanley, D.F. Spontaneous intracerebral hemorrhage. N. Engl. J. Med. 2001, 344, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Y.; Betz, A.L.; Chenevert, T.L.; Brunberg, J.A.; Hoff, J.T. Experimental intracerebral hemorrhage: Relationship between brain edema, blood flow, and blood-brain barrier permeability in rats. J. Neurosurg. 1994, 1, 93–102. [Google Scholar] [CrossRef]
- Katsuki, H. Exploring neuroprotective drug therapies for intracerebral hemorrhage. J. Pharmacol. Sci. 2010, 4, 366–378. [Google Scholar] [CrossRef]
- Keep, R.F.; Xiang, J.; Ennis, S.R.; Andjelkovic, A.; Hua, Y.; Xi, G.; Hoff, J.T. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir. Suppl. 2008, 105, 73–77. [Google Scholar] [PubMed]
- Keep, R.F.; Zhou, N.; Xiang, J.; Andjelkovic, A.V.; Hua, Y.; Xi, G. Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS 2014, 10, 18. [Google Scholar] [CrossRef]
- Kreiter, K.T.; Copeland, D.; Bernardini, G.L.; Bates, J.E.; Peery, S.; Claassen, J.; Du, Y.E.; Stern, Y.; Connolly, E.S.; Mayer, S.A. Predictors of cognitive dysfunction after subarachnoid hemorrhage. Stroke 2002, 1, 200–208. [Google Scholar] [CrossRef]
- Bodmer, D.; Vaughan, K.A.; Zacharia, B.E.; Hickman, Z.L.; Connolly, E.S. The Molecular Mechanisms that Promote Edema after Intracerebral Hemorrhage. Transl. Stroke Res. 2012, 3, S52–S61. [Google Scholar] [CrossRef]
- Kirkman, M.A.; Allan, S.M.; Parry-Jones, A.R. Experimental intracerebral hemorrhage: Avoiding pitfalls in translational research. J. Cereb. Blood Flow Metab. 2011, 11, 2135–2151. [Google Scholar] [CrossRef]
- Manaenko, A.; Chen, H.; Zhang, J.H.; Tang, J. Comparison of different preclinical models of intracerebral hemorrhage. Acta Neurochir. Suppl. 2011, 111, 9–14. [Google Scholar] [PubMed]
- Kooijman, E.; Nijboer, C.H.; van Velthoven, C.T.; Kavelaars, A.; Kesecioglu, J.; Heijnen, C.J. The rodent endovascular puncture model of subarachnoid hemorrhage: Mechanisms of brain damage and therapeutic strategies. J. Neuroinflamm. 2014, 11, 2. [Google Scholar] [CrossRef]
- Carpenter, J.; Weinstein, S.; Myseros, J.; Vezina, G.; Bell, M.J. Inadvertent hyponatremia leading to acute cerebral edema and early evidence of herniation. Neurocrit. Care 2007, 3, 195–199. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin-4 and brain edema. Pediatr. Nephrol. 2007, 6, 778–784. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Wu, S.; Ehara, K.; Nagashima, T.; Tamaki, N. Cerebral blood flow of rats with water-intoxicated brain edema. Acta Neurochir. Suppl. (Wien) 1994, 60, 190–192. [Google Scholar]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zador, Z.; Verkman, A.S. Glial cell aquaporin-4 overexpression in transgenic mice accelerates cytotoxic brain swelling. J. Biol. Chem. 2008, 22, 15280–15286. [Google Scholar] [CrossRef]
- Yeung, P.K.; Lo, A.C.; Leung, J.W.; Chung, S.S.; Chung, S.K. Targeted overexpression of endothelin-1 in astrocytes leads to more severe cytotoxic brain edema and higher mortality. J. Cereb. Blood Flow Metab. 2009, 12, 1891–1902. [Google Scholar] [CrossRef]
- Kozler, P.; Riljak, V.; Pokorný, J. Both water intoxication and osmotic BBB disruption increase brain water content in rats. Physiol. Res. 2013, 62, S75–S80. [Google Scholar] [PubMed]
- Mpabanzi, L.; Jalan, R. Neurological complications of acute liver failure: Pathophysiological basis of current management and emerging therapies. Neurochem. Int. 2012, 60, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D. Low grade cerebral edema and the pathogenesis of hepatic encephalopathy in cirrhosis. Hepatology 2006, 43, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.S.; Wendon, J. Brain edema in liver failure: Basic physiologic principles and management. Liver Transpl. 2002, 8, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Wendon, J.; Lee, W. Encephalopathy and cerebral edema in the setting of acute liver failure: Pathogenesis and management. Neurocrit. Care 2008, 1, 97–102. [Google Scholar] [CrossRef]
- Scott, T.R.; Kronsten, V.T.; Hughes, R.D.; Shawcross, D.L. Pathophysiology of cerebral oedema in acute liver failure. World J. Gastroenterol. 2013, 48, 9240–9255. [Google Scholar] [CrossRef]
- Larsen, F.S.; Wendon, J. Prevention and management of brain edema in patients with acute liver failure. Liver Transpl. 2008, 14, S90–S96. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Davies, N.A.; Shawcross, D.L.; Hodges, S.J.; Zwingmann, C.; Brooks, H.F.; Mani, A.R.; Harry, D.; Stadlbauer, V.; Zou, Z.; et al. Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology 2007, 45, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Swain, M.; Butterworth, R.F.; Blei, A.T. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992, 15, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Rao, K.V.; Jayakumar, A.R. Mechanisms of ammonia-induced astrocyte swelling. Metab. Brain Dis. 2005, 20, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Verkman, A.S.; Curtis, K.M.; Norenberg, M.D. Aquaporin-4 deletion in mice reduces encephalopathy and brain edema in experimental acute liver failure. Neurobiol. Dis. 2014, 63, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Rao, K.V.; Murthy, C.R.; Norenberg, M.D. Glutamine in the mechanism of ammonia-induced astrocyte swelling. Neurochem. Int. 2006, 6–7, 623–628. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Jayakumar, A.R.; Norenberg, M.D. Glutamine in the pathogenesis of acute hepatic encephalopathy. Neurochem. Int. 2012, 4, 575–580. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Bethea, J.R.; Tong, X.Y.; Gomez, J.; Norenberg, M.D. NF-κB in the mechanism of brain edema in acute liver failure: Studies in transgenic mice. Neurobiol. Dis. 2011, 2, 498–507. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Valdes, V.; Tong, X.Y.; Shamaladevi, N.; Gonzalez, W.; Norenberg, M.D. Sulfonylurea receptor 1 contributes to the astrocyte swelling and brain edema in acute liver failure. Transl. Stroke Res. 2014, 1, 28–37. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Jayakumar, A.R.; Tong, X.; Curtis, K.M.; Norenberg, M.D. Brain aquaporin-4 in experimental acute liver failure. J. Neuropathol. Exp. Neurol. 2010, 9, 869–879. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Reddy, P.V.; Tong, X.; Norenberg, M.D. Brain edema in acute liver failure: Inhibition by l-histidine. Am. J. Pathol. 2010, 3, 1400–1408. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Jayakumar, A.R.; Norenberg, M.D. Brain edema in acute liver failure: Mechanisms and concepts. Metab. Brain Dis. 2014, 4, 927–936. [Google Scholar] [CrossRef]
- Cauli, O.; López-Larrubia, P.; Rodrigo, R.; Agusti, A.; Boix, J.; Nieto-Charques, L.; Cerdán, S.; Felipo, V. Brain region-selective mechanisms contribute to the progression of cerebral alterations in acute liver failure in rats. Gastroenterology 2011, 140, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Nguyen, J.H. TIMP-1/MMP-9 imbalance in brain edema in rats with fulminant hepatic failure. J. Surg. Res. 2006, 134, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Chavarria, L.; Cordoba, J. Magnetic resonance of the brain in chronic and acute liver failure. Metab. Brain Dis. 2014, 29, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Chen, M.; Simard, J.M.; Norenberg, M.D. Increased aquaporin-4 expression in ammonia-treated cultured astrocytes. Neuroreport 2003, 18, 2379–2382. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Liu, M.; Moriyama, M.; Ramakrishnan, R.; Forbush, B., 3rd; Reddy, P.V.; Norenberg, M.D. Na–K–Cl cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J. Biol. Chem. 2008, 49, 33874–33882. [Google Scholar] [CrossRef]
- Keep, R.F.; Hua, Y.; Xi, G. Brain water content. A misunderstood measurement? Transl. Stroke Res. 2012, 3, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, A.; Poll, W.; Shulman, K.; Bhagavan, H. A simple gravimetric technique for measurement of cerebral edema. J. Neurosurg. 1978, 49, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Loubinoux, I.; Volk, A.; Borredon, J.; Guirimand, S.; Tiffon, B.; Seylaz, J.; Méric, P. Spreading of vasogenic edema and cytotoxic edema assessed by quantitative diffusion and T2 magnetic resonance imaging. Stroke 1997, 28, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Ashwal, S.; Tone, B.; Regli, L.; Tian, H.R.; Obenaus, A. Temporal and regional evolution of aquaporin-4 expression and magnetic resonance imaging in a rat pup model of neonatal stroke. Pediatr. Res. 2007, 62, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Rumpel, H.; Ferrini, B.; Martin, E. Lasting cytotoxic edema as an indicator of irreversible brain damage: A case of neonatal stroke. AJNR Am. J. Neuroradiol. 1998, 19, 1636–1638. [Google Scholar] [PubMed]
- Roy, H.; Bhardwaj, S.; Ylä-Herttuala, S. Biology of vascular endothelial growth factors. FEBS Lett. 2006, 12, 2879–2887. [Google Scholar] [CrossRef]
- Hayashi, T.; Abe, K.; Suzuki, H.; Itoyama, Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke 1997, 28, 2039–2044. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Eskandarian, M.R.; Davis, J.; Eubanks, J.H. Differential expression of vascular endothelial growth factor-A (VEGF-A) and VEGF-B after brain injury. J. Neuropathol. Exp. Neurol. 2002, 9, 778–788. [Google Scholar]
- Sköld, M.K.; von Gertten, C.; Sandberg-Nordqvist, A.C.; Mathiesen, T.; Holmin, S. VEGF and VEGF receptor expression after experimental brain contusion in rat. J. Neurotrauma 2005, 22, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Issa, R.; Krupinski, J.; Bujny, T.; Kumar, S.; Kaluza, J.; Kumar, P. Vascular endothelial growth factor and its receptor, KDR, in human brain tissue after ischemic stroke. Lab. Investig. 1999, 4, 417–425. [Google Scholar]
- Plate, K.H.; Beck, H.; Danner, S.; Allegrini, P.R.; Wiessner, C. Cell type specific upregulation of vascular endothelial growth factor in an MCA-occlusion model of cerebral infarct. J. Neuropathol. Exp. Neurol. 1999, 6, 654–666. [Google Scholar] [CrossRef]
- Mărgăritescu, O.; Pirici, D.; Mărgăritescu, C. VEGF expression in human brain tissue after acute ischemic stroke. Rom. J. Morphol. Embryol. 2011, 4, 1283–1292. [Google Scholar]
- Shore, P.M.; Jackson, E.K.; Wisniewski, S.R.; Clark, R.S.; Adelson, P.D.; Kochanek, P.M. Vascular endothelial growth factor is increased in cerebrospinal fluid after traumatic brain injury in infants and children. Neurosurgery 2004, 54, 605–612. [Google Scholar] [CrossRef]
- Hirose, T.; Matsumoto, N.; Tasaki, O.; Nakamura, H.; Akagaki, F.; Shimazu, T. Delayed progression of edema formation around a hematoma expressing high levels of VEGF and MMP-9 in a patient with traumatic brain injury: Case report. Neurol. Med. Chir. (Tokyo) 2013, 9, 609–612. [Google Scholar] [CrossRef]
- Matsuo, R.; Ago, T.; Kamouchi, M.; Kuroda, J.; Kuwashiro, T.; Hata, J.; Sugimori, H.; Fukuda, K.; Gotoh, S.; Makihara, N.; et al. Clinical significance of plasma VEGF value in ischemic stroke-research for biomarkers in ischemic stroke (REBIOS) study. BMC Neurol. 2013, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Bell, J.D.; Siddiq, I.P.; Baker, A.J. An analysis of regional microvascular loss and recovery following two grades of fluid percussion trauma: A role for hypoxia-inducible factors in traumatic brain injury. J. Cereb. Blood Flow Metab. 2009, 3, 575–584. [Google Scholar] [CrossRef]
- Lee, C.; Agoston, D.V. Vascular endothelial growth factor is involved in mediating increased de novo hippocampal neurogenesis in response to traumatic brain injury. J. Neurotrauma 2010, 3, 541–553. [Google Scholar] [CrossRef]
- Jośko, J. Cerebral angiogenesis and expression of VEGF after subarachnoid hemorrhage (SAH) in rats. Brain Res. 2003, 1–2, 58–69. [Google Scholar] [CrossRef]
- Li, Y.N.; Pan, R.; Qin, X.J.; Yang, W.L.; Qi, Z.; Liu, W.; Liu, K.J. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J. Neurochem. 2014, 1, 120–129. [Google Scholar] [CrossRef]
- Bates, D.O. Vascular endothelial growth factors and vascular permeability. Cardiovasc. Res. 2010, 87, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 7, 2454–2468. [Google Scholar] [CrossRef]
- Jiang, S.; Xia, R.; Jiang, Y.; Wang, L.; Gao, F. Vascular endothelial growth factors enhance the permeability of the mouse blood-brain barrier. PLoS ONE 2014, 2, e86407. [Google Scholar] [CrossRef]
- Tsukita, S.; Furuse, M. Claudin-based barrier in simple and stratified cellular sheets. Curr. Opin. Cell Biol. 2002, 14, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; Nakase, H.; Tamaki, R.; Sakaki, T. Vascular endothelial growth factor antagonist reduces brain edema formation and venous infarction. Stroke 2005, 36, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Van Bruggen, N.; Thibodeaux, H.; Palmer, J.T.; Lee, W.P.; Fu, L.; Cairns, B.; Tumas, D.; Gerlai, R.; Williams, S.P.; van Lookeren, C.M.; et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J. Clin. Investig. 1999, 104, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Kumai, Y.; Ooboshi, H.; Ibayashi, S.; Ishikawa, E.; Sugimori, H.; Kamouchi, M.; Kitazono, T.; Egashira, K.; Iida, M. Postischemic gene transfer of soluble Flt-1 protects against brain ischemia with marked attenuation of blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2007, 6, 1152–1160. [Google Scholar] [CrossRef]
- Koyama, J.; Miyake, S.; Sasayama, T.; Kondoh, T.; Kohmura, E. Effect of VEGF receptor antagonist (VGA1155) on brain edema in the rat cold injury model. Kobe J. Med. Sci. 2007, 5, 199–207. [Google Scholar]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 2004, 5, 558–564. [Google Scholar] [CrossRef]
- Pepper, M.S. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1104–1117. [Google Scholar] [CrossRef]
- Seo, J.H.; Guo, S.; Lok, J.; Navaratna, D.; Whalen, M.J.; Kim, K.W.; Lo, E.H. Neurovascular matrix metalloproteinases and the blood-brain barrier. Curr. Pharm. Des. 2012, 25, 3645–3648. [Google Scholar] [CrossRef]
- Heo, J.H.; Lucero, J.; Abumiya, T.; Koziol, J.A.; Copeland, B.R.; del Zoppo, G.J. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1999, 6, 624–633. [Google Scholar] [CrossRef]
- Grossetete, M.; Phelps, J.; Arko, L.; Yonas, H.; Rosenberg, G.A. Elevation of matrix metalloproteinases 3 and 9 in cerebrospinal fluid and blood in patients with severe traumatic brain injury. Neurosurgery 2009, 4, 702–708. [Google Scholar] [CrossRef]
- Zheng, K.; Li, C.; Shan, X.; Liu, H.; Fan, W.; Wang, Z.; Zheng, P. Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with moderate and severe traumatic brain injury. Neurol. India 2013, 6, 606–609. [Google Scholar]
- Jia, F.; Pan, Y.H.; Mao, Q.; Liang, Y.M.; Jiang, J.Y. Matrix metalloproteinase-9 expression and protein levels after fluid percussion injury in rats: The effect of injury severity and brain temperature. J. Neurotrauma 2010, 6, 1059–1068. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Navratil, M. Metalloproteinase inhibition blocks edema in intracerebral hemorrhage in the rat. Neurology 1997, 4, 921–926. [Google Scholar] [CrossRef]
- Planas, A.M.; Solé, S.; Justicia, C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol. Dis. 2001, 8, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A.; Cunningham, L.A.; Wallace, J.; Alexander, S.; Estrada, E.Y.; Grossetete, M.; Razhagi, A.; Miller, K.; Gearing, A. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: Activation of MMP9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001, 893, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Muir, E.M.; Adcock, K.H.; Morgenstern, D.A.; Clayton, R.; von Stillfried, N.; Rhodes, K.; Ellis, C.; Fawcett, J.W.; Rogers, J.H. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res. Mol. Brain Res. 2002, 100, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Tsuji, K.; Lee, S.R.; Lo, E.H. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J. Neurosci. 2004, 3, 671–678. [Google Scholar] [CrossRef]
- Morita-Fujimura, Y.; Fujimura, M.; Gasche, Y.; Copin, J.C.; Chan, P.H. Overexpression of copper and zinc superoxide dismutase in transgenic mice prevents the induction and activation of matrix metalloproteinases after cold injury-induced brain trauma. J. Cereb. Blood Flow Metab. 2000, 1, 130–138. [Google Scholar] [CrossRef]
- Gasche, Y.; Copin, J.C.; Sugawara, T.; Fujimura, M.; Chan, P.H. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2001, 21, 1393–1400. [Google Scholar] [CrossRef]
- Rivera, S.; Ogier, C.; Jourquin, J.; Timsit, S.; Szklarczyk, A.W.; Miller, K.; Gearing, A.J.; Kaczmarek, L.; Khrestchatisky, M. Gelatinase B and TIMP-1 are regulated in a cell- and time-dependent manner in association with neuronal death and glial reactivity after global forebrain ischemia. Eur. J. Neurosci. 2002, 15, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Asahi, M.; Wang, X.; Mori, T.; Sumii, T.; Jung, J.C.; Moskowitz, M.A.; Fini, M.E.; Lo, E.H. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 2001, 21, 7724–7732. [Google Scholar] [PubMed]
- Wang, J.; Tsirka, S.E. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain 2005, 128, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Shigemori, Y.; Katayama, Y.; Mori, T.; Maeda, T.; Kawamata, T. Matrix metalloproteinase-9 is associated with blood-brain barrier opening and brain edema formation after cortical contusion in rats. Acta Neurochir. Suppl. 2006, 96, 130–133. [Google Scholar] [PubMed]
- Rosenberg, G.A.; Estrada, E.Y.; Dencoff, J.E. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke 1998, 10, 2189–2195. [Google Scholar] [CrossRef]
- Sood, R.R.; Taheri, S.; Candelario-Jalil, E.; Estrada, E.Y.; Rosenberg, G.A. Early beneficial effect of matrix metalloproteinase inhibition on blood-brain barrier permeability as measured by magnetic resonance imaging countered by impaired long-term recovery after stroke in rat brain. J. Cereb. Blood Flow Metab. 2008, 28, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Kawai, N.; Kawanishi, M.; Okada, M.; Matsumoto, Y.; Nagao, S. Treatment of cold injury-induced brain edema with a nonspecific matrix metalloproteinase inhibitor MMI270 in rats. J. Neurotrauma 2003, 7, 649–657. [Google Scholar] [CrossRef]
- Agre, P.; King, L.S.; Yasui, M.; Guggino, W.B.; Ottersen, O.P.; Fujiyoshi, Y.; Engel, A.; Nielsen, S. Aquaporin water channels—From atomic structure to clinical medicine. J. Physiol. 2002, 1, 3–16. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 4, 265–277. [Google Scholar] [CrossRef]
- Fukuda, A.M.; Badaut, J. Aquaporin 4: A player in cerebral edema and neuroinflammation. J. Neuroinflamm. 2012, 9, 279. [Google Scholar] [CrossRef]
- Oshio, K.; Watanabe, H.; Song, Y.; Verkman, A.S.; Manley, G.T. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel Aquaporin-1. FASEB J. 2005, 19, 76–78. [Google Scholar] [PubMed]
- Badaut, J.; Hirt, L.; Granziera, C.; Bogousslavsky, J.; Magistretti, P.J.; Regli, L. Astrocyte-specific expression of aquaporin-9 in mouse brain is increased after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2001, 21, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Yao, H.T.; Zhang, W.P.; Zhang, L.; Ding, W.; Zhang, S.H.; Chen, Z.; Wei, E.Q. Increased expression of aquaporin-4 in human traumatic brain injury and brain tumors. J. Zhejiang Univ. Sci. B 2005, 1, 33–37. [Google Scholar] [CrossRef]
- Kimbler, D.E.; Shields, J.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice. PLoS ONE 2012, 7, e41229. [Google Scholar] [CrossRef] [PubMed]
- Qing, W.G.; Dong, Y.Q.; Ping, T.Q.; Lai, L.G.; Fang, L.D.; Min, H.W.; Xia, L.; Heng, P.Y. Brain edema after intracerebral hemorrhage in rats: The role of iron overload and aquaporin 4. J. Neurosurg. 2009, 3, 462–468. [Google Scholar] [CrossRef]
- Kiening, K.L.; van Landeghem, F.K.; Schreiber, S.; Thomale, U.W.; von Deimling, A.; Unterberg, A.W.; Stover, J.F. Decreased hemispheric Aquaporin-4 is linked to evolving brain edema following controlled cortical impact injury in rats. Neurosci. Lett. 2002, 2, 105–108. [Google Scholar] [CrossRef]
- Hoshi, A.; Yamamoto, T.; Shimizu, K.; Sugiura, Y.; Ugawa, Y. Chemical preconditioning-induced reactive astrocytosis contributes to the reduction of post-ischemic edema through aquaporin-4 downregulation. Exp. Neurol. 2011, 1, 89–95. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Manley, G.T.; Krishna, S.; Verkman, A.S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004, 18, 1291–1293. [Google Scholar] [PubMed]
- Tang, Y.; Wu, P.; Su, J.; Xiang, J.; Cai, D.; Dong, Q. Effects of A quaporin-4 on edema formation following intracerebral hemorrhage. Exp. Neurol. 2010, 2, 485–495. [Google Scholar] [CrossRef]
- Igarashi, H.; Huber, V.J.; Tsujita, M.; Nakada, T. Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol. Sci. 2011, 32, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Pandey, A.K.; Paul, S.; Patnaik, R.; Yavagal, D.R. Aquaporin-4 inhibition mediates piroxicam-induced neuroprotection against focal cerebral ischemia/reperfusion injury in rodents. PLoS ONE 2013, 8, e73481. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Norenberg, M.D. The Na–K–Cl co-transporter in astrocyte swelling. Metab. Brain Dis. 2010, 1, 31–38. [Google Scholar] [CrossRef]
- Yan, Y.; Dempsey, R.J.; Flemmer, A.; Forbush, B.; Sun, D. Inhibition of Na+–K+–Cl− cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003, 1, 22–31. [Google Scholar] [CrossRef]
- Simard, J.M.; Kahle, K.T.; Gerzanich, V. Molecular mechanisms of microvascular failure in CNS injury—Synergistic roles of NKCC1 and SUR1/TRPM4. J. Neurosurg. 2010, 3, 622–629. [Google Scholar] [CrossRef]
- Chechneva, O.; Yuen, N.; Tsai, Y.-C.; Chen, Y.J.; Anderson, S.; O’Donnell, M. Evidence for blood-brain barrier Na–K–Cl cotransport, Na/H exchange and Na–HCO3 cotransport involvement in hyperglycemia exacerbation of cerebral edema formation in ischemic stroke. FASEB J. 2014, 28, S685.3. [Google Scholar]
- Su, G.; Kintner, D.B.; Flagella, M.; Shull, G.E.; Sun, D. Astrocytes from Na+–K+–Cl− cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am. J. Physiol. Cell Physiol. 2002, 5, C1147–C1160. [Google Scholar] [CrossRef]
- Chen, H.; Luo, J.; Kintner, D.B.; Shull, G.E.; Sun, D. Na+-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2005, 1, 54–66. [Google Scholar] [CrossRef]
- O’Donnell, M.E.; Tran, L.; Lam, T.I.; Liu, X.B.; Anderson, S.E. Bumetanide inhibition of the blood-brain barrier Na–K–Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J. Cereb. Blood Flow Metab. 2004, 9, 1046–1056. [Google Scholar] [CrossRef]
- Lu, K.T.; Cheng, N.C.; Wu, C.Y.; Yang, Y.L. NKCC1-mediated traumatic brain injury-induced brain edema and neuron death via Raf/MEK/MAPK cascade. Crit. Care Med. 2008, 3, 917–922. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Valdes, V.; Norenberg, M.D. The Na–K–Cl cotransporter in the brain edema of acute liver failure. J. Hepatol. 2011, 2, 272–278. [Google Scholar] [CrossRef]
- Chen, M.; Simard, J.M. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J. Neurosci. 2001, 17, 6512–6521. [Google Scholar]
- Simard, J.M.; Chen, M.; Tarasov, K.V.; Bhatta, S.; Ivanova, S.; Melnitchenko, L.; Tsymbalyuk, N.; West, G.A.; Gerzanich, V. Newly expressed SUR1-regulated NCCa-ATP channel mediates cerebral edema after ischemic stroke. Nat. Med. 2006, 4, 433–440. [Google Scholar] [CrossRef]
- Simard, J.M.; Woo, S.K.; Schwartzbauer, G.T.; Gerzanich, V. Sulfonylurea receptor 1 in central nervous system injury: A focused review. J. Cereb. Blood Flow Metab. 2012, 9, 699–717. [Google Scholar]
- Simard, J.M.; Tsymbalyuk, O.; Ivanov, A.; Ivanova, S.; Bhatta, S.; Geng, Z.; Woo, S.K.; Gerzanich, V. Endothelial sulfonylurea receptor 1-regulated NCCa-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J. Clin. Investig. 2007, 8, 2105–2013. [Google Scholar] [CrossRef]
- Zweckberger, K.; Hackenberg, K.; Jung, C.S.; Hertle, D.N.; Kiening, K.L.; Unterberg, A.W.; Sakowitz, O.W. Glibenclamide reduces secondary brain damage after experimental traumatic brain injury. Neuroscience 2014, 272, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Woo, S.K.; Tsymbalyuk, N.; Voloshyn, O.; Yurovsky, V.; Ivanova, S.; Lee, R.; Gerzanich, V. Glibenclamide-10-h treatment window in a clinically relevant model of stroke. Transl. Stroke Res. 2012, 2, 286–295. [Google Scholar] [CrossRef]
- Simard, J.M.; Sheth, K.N.; Kimberly, W.T.; Stern, B.J.; del Zoppo, G.J.; Jacobson, S.; Gerzanich, V. Glibenclamide in cerebral ischemia and stroke. Neurocrit. Care 2014, 2, 319–333. [Google Scholar] [CrossRef]
- Goto, K.; Hama, H.; Kasuya, Y. Molecular pharmacology and pathophysiological significance of endothelin. Jpn. J. Pharmacol. 1996, 72, 261–290. [Google Scholar] [CrossRef] [PubMed]
- Schinelli, S. Pharmacology and physiopathology of the brain endothelin system: An overview. Curr. Med. Chem. 2006, 13, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Dashwood, M.R.; Loesch, A. Endothelin-1 as a neuropeptide: Neurotransmitter or neurovascular effects? J. Cell Commun. Signal. 2010, 4, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Kaundal, R.K.; Deshpande, T.A.; Gulati, A.; Sharma, S.S. Targeting endothelin receptors for pharmacotherapy of ischemic stroke: Current scenario and future perspectives. Drug Discov. Today 2012, 17, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.M.; Rogers, S.D.; Pomonis, J.D.; Egnaczyk, G.F.; Keyser, C.P.; Schmidt, J.A.; Ghilardi, J.R.; Maggio, J.E.; Mantyh, P.W. Endothelin receptor expression in the normal and injured spinal cord: Potential involvement in injury-induced ischemia and gliosis. Exp. Neurol. 2003, 180, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.D.; Peters, C.M.; Pomonis, J.D.; Hagiwara, H.; Ghilardi, J.R.; Mantyh, P.W. Endothelin B receptors are expressed by astrocytes and regulate astrocyte hypertrophy in the normal and injured CNS. Glia 2003, 41, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsson, U.; Li, L.; Pekna, M.; Berthold, C.H.; Blom, S.; Eliasson, C.; Renner, O.; Bushong, E.; Ellisman, M.; Morgan, T.E.; et al. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J. Neurosci. 2004, 24, 5016–5021. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Takemura, M.; Koyama, Y.; Shigenaga, Y.; Okada, T.; Baba, A. Endothelins promote the activation of astrocytes in rat neostriatum through ETB receptors. Eur. J. Neurosci. 1997, 9, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Tsujikawa, K.; Matsuda, T.; Baba, A. Intracerebroventricular administration of an endothelin ETB receptor agonist increases expressions of GDNF and BDNF in rat brain. Eur. J. Neurosci. 2003, 18, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Tanaka, K. Intracerebroventricular administration of an endothelin ETB-receptor agonist increases expression of matrix metalloproteinase-2 and -9 in rat brain. J. Pharmacol. Sci. 2010, 114, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Nagae, R.; Tokuyama, S.; Tanaka, K. I.c.v administration of an endothelin ETB receptor agonist stimulates vascular endothelial growth factor-A production and activates vascular endothelial growth factor receptors in rat brain. Neuroscience 2011, 192, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Maebara, Y.; Hayashi, M.; Nagae, R.; Tokuyama, S.; Michinaga, S. Endothelins reciprocally regulate VEGF-A and angiopoietin-1 production in cultured rat astrocytes: Implications on astrocytic proliferation. Glia 2012, 60, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Michinaga, S.; Nagase, M.; Matsuyama, E.; Yamanaka, D.; Seno, N.; Fuka, M.; Yamamoto, Y.; Koyama, Y. Amelioration of cold injury-induced cortical brain edema formation by selective endothelin ETB receptor antagonists in mice. PLoS ONE 2014, 9, e102009. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Ryu, H.J.; Kang, T.C. Status epilepticus induces vasogenic edema via tumor necrosis factor-α/endothelin-1-mediated two different pathways. PLoS ONE 2013, 9, e74458. [Google Scholar] [CrossRef]
- Moldes, O.; Sobrino, T.; Blanco, M.; Agulla, J.; Barral, D.; Ramos-Cabrer, P.; Castillo, J. Neuroprotection afforded by antagonists of endothelin-1 receptors in experimental stroke. Neuropharmacology 2012, 63, 279–285. [Google Scholar] [CrossRef]
- Kaal, E.C.; Vecht, C.J. The management of brain edema in brain tumors. Curr. Opin. Oncol. 2004, 6, 593–600. [Google Scholar] [CrossRef]
- Sinha, S.; Bastin, M.E.; Wardlaw, J.M.; Armitage, P.A.; Whittle, I.R. Effects of dexamethasone on peritumoral oedematous brain: A DT-MRI study. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1632–1635. [Google Scholar] [CrossRef] [PubMed]
- Betz, A.L.; Coester, H.C. Effect of steroids on edema and sodium uptake of the brain during focal ischemia in rats. Stroke 1990, 8, 1199–1204. [Google Scholar] [CrossRef]
- Hortobágyi, T.; Hortobágyi, S.; Görlach, C.; Harkany, T.; Benyó, Z.; Görögh, T.; Nagel, W.; Wahl, M. A novel brain trauma model in the mouse: Effects of dexamethasone treatment. Pflugers Arch. 2000, 2–3, 409–415. [Google Scholar]
- Vachon, P.; Moreau, J.P. Low doses of dexamethasone decrease brain water content of collagenase-induced cerebral hematoma. Can. J. Vet. Res. 2003, 2, 157–159. [Google Scholar]
- Yang, J.T.; Lee, T.H.; Lee, I.N.; Chung, C.Y.; Kuo, C.H.; Weng, H.H. Dexamethasone inhibits ICAM-1 and MMP-9 expression and reduces brain edema in intracerebral hemorrhagic rats. Acta Neurochir. (Wien) 2011, 11, 2197–2203. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Dimitrijevic, O.B.; Keep, R.F.; Andjelkovic, A.V. Inflammation and brain edema: New insights into the role of chemokines and their receptors. Acta Neurochir. Suppl. 2006, 96, 444–450. [Google Scholar] [PubMed]
- Kim, H.; Lee, J.M.; Park, J.S.; Jo, S.A.; Kim, Y.O.; Kim, C.W.; Jo, I. Dexamethasone coordinately regulates angiopoietin-1 and VEGF: A mechanism of glucocorticoid-induced stabilization of blood-brain barrier. Biochem. Biophys. Res. Commun. 2008, 1, 243–248. [Google Scholar] [CrossRef]
- Romero, I.A.; Radewicz, K.; Jubin, E.; Michel, C.C.; Greenwood, J.; Couraud, P.O.; Adamson, P. Changes in cytoskeletal and tight junctional proteins correlate with decreased permeability induced by dexamethasone in cultured rat brain endothelial cells. Neurosci. Lett. 2003, 2, 112–116. [Google Scholar] [CrossRef]
- Kimberly, W.T.; Battey, T.W.; Pham, L.; Wu, O.; Yoo, A.J.; Furie, K.L.; Singhal, A.B.; Elm, J.J.; Stern, B.J.; Sheth, K.N. Glyburide is associated with attenuated vasogenic edema in stroke patients. Neurocrit. Care 2014, 2, 193–201. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michinaga, S.; Koyama, Y. Pathogenesis of Brain Edema and Investigation into Anti-Edema Drugs. Int. J. Mol. Sci. 2015, 16, 9949-9975. https://doi.org/10.3390/ijms16059949
Michinaga S, Koyama Y. Pathogenesis of Brain Edema and Investigation into Anti-Edema Drugs. International Journal of Molecular Sciences. 2015; 16(5):9949-9975. https://doi.org/10.3390/ijms16059949
Chicago/Turabian StyleMichinaga, Shotaro, and Yutaka Koyama. 2015. "Pathogenesis of Brain Edema and Investigation into Anti-Edema Drugs" International Journal of Molecular Sciences 16, no. 5: 9949-9975. https://doi.org/10.3390/ijms16059949