
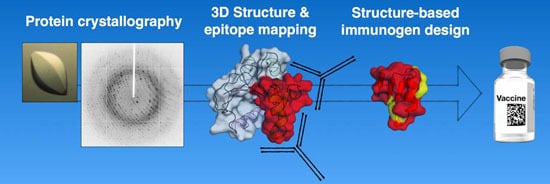
Protein Crystallography in Vaccine Research and Development

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Protein Crystallography for Antigen Characterization and Epitope Mapping
2.1. Antigen Characterization by X-ray Crystallography
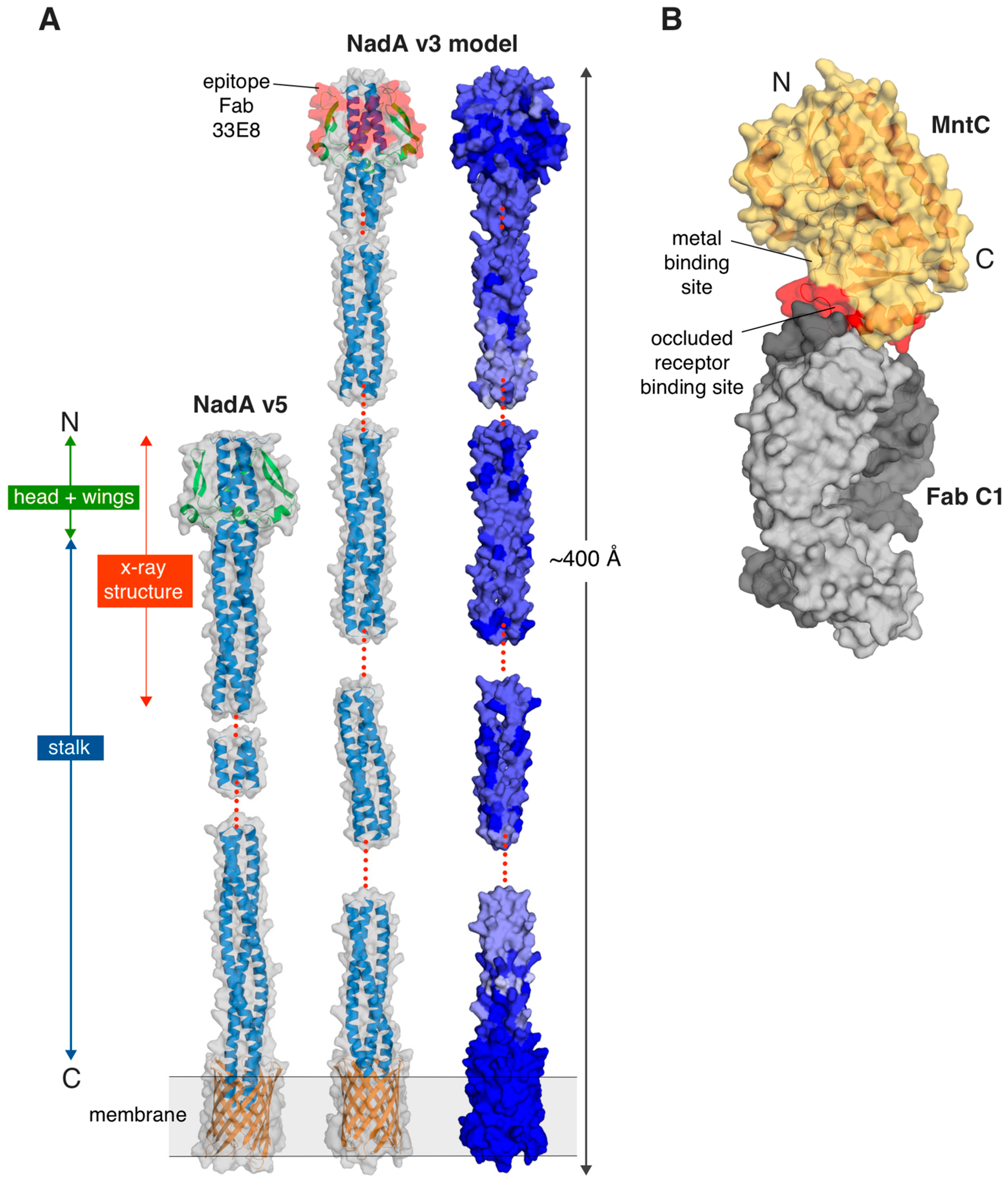
2.1.1. NadA—A Surface-Exposed Meningococcal Adhesin and Vaccine Antigen

2.1.2. Staphylococcal Solute Binding Protein Antigens
2.2. Protein Crystallography and High-Resolution B-Cell Epitope Mapping

3. Structure-Based Antigen Design
3.1. Optimizing the Factor H Binding Protein Antigen of Serogroup B Meningococcus
3.1.1. Overcoming Sequence Variability
3.1.2. Elimination of Undesirable Function
3.2. Nonbinding Mutants of Transferrin Binding Protein B
3.3. Multiple Protein F-Based Strategies for a Respiratory Syncytial Virus Vaccine
3.3.1. Rational Engineering of a Soluble, Stable and Homogeneous Post-Fusion F
3.3.2. An Antibody-Dependent Approach to Design and Engineer Pre-Fusion F

3.3.3. Epitope-Focused Vaccine Design to Target a Neutralizing Epitope on the F Antigen
3.4. Human Immunodeficiency Virus (HIV)—The Ultimate Challenge?
3.4.1. Scaffold-Based and Multi-Copy Approaches in the Design of HIV Antigens
3.4.2. Structure Determinations of the HIV Envelope Glycoprotein (Env) Trimer
4. Enabling Technology for Protein Crystallography in Vaccine Research
4.1. A Short Introduction to X-ray Crystallography of Proteins
4.2. Advances in Protein Crystallization
4.2.1. Group 1
- Surface entropy reduction (SER): mutation of surface residues to create patches of low entropy that can preferentially mediate crystal contacts [128];
- Sequence homolog screenings: sequence variability between homolog proteins, if localized on the surface, can favor better packing and crystallization [21];
4.2.2. Group 2
- Binding partners (or chaperone-assisted crystallography): Fab fragments, single-domain antibodies, synthetic antibodies, and more general substrates, may reduce conformational freedom of the target protein and thus enhance propensity to form the ordered lattice required for crystallization [118,131,132];
- In situ proteolysis: the addition of trace amounts of proteases in crystallization trials can help to enzymatically eliminate flexible or disordered regions that might hinder crystallization [133];
4.3. Advances Facilitating the Determination of Crystal Structures
5. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Plotkin, S. History of vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 12283–12287. [Google Scholar] [CrossRef] [PubMed]
- Whitney, C.G.; Zhou, F.; Singleton, J.; Schuchat, A. Benefits from immunization during the vaccines for children program era—United States, 1994–2013. Morb. Mortal. Wkly. Rep. 2014, 63, 352–355. [Google Scholar]
- Delany, I.; Rappuoli, R.; de Gregorio, E. Vaccines for the 21st century. EMBO Mol. Med. 2014, 6, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Reverse vaccinology, a genome-based approach to vaccine development. Vaccine 2001, 19, 2688–2691. [Google Scholar] [CrossRef]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B.; et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, M.M.; Adu-Bobie, J.; Comanducci, M.; Arico, B.; Savino, S.; Santini, L.; Brunelli, B.; Bambini, S.; Biolchi, A.; Capecchi, B.; et al. A universal vaccine for serogroup B meningococcus. Proc. Natl. Acad. Sci. USA 2006, 103, 10834–10839. [Google Scholar] [CrossRef] [PubMed]
- Pace, D.; Pollard, A.J. Meningococcal disease: Clinical presentation and sequelae. Vaccine 2012, 30 (Suppl. 2), B3–B9. [Google Scholar] [CrossRef] [PubMed]
- Pace, D. Quadrivalent meningococcal ACYW-135 glycoconjugate vaccine for broader protection from infancy. Expert Rev. Vaccines 2009, 8, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Finne, J.; Bitter-Suermann, D.; Goridis, C.; Finne, U. An IgG monoclonal antibody to group B meningococci cross-reacts with developmentally regulated polysialic acid units of glycoproteins in neural and extraneural tissues. J. Immunol. 1987, 138, 4402–4407. [Google Scholar] [PubMed]
- O’Ryan, M.; Stoddard, J.; Toneatto, D.; Wassil, J.; Dull, P.M. A multi-component meningococcal serogroup B vaccine (4CMenB): The clinical development program. Drugs 2014, 74, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, G.W.; Jones, T.R.; Liberator, P.; Hao, L.; Harris, S.; McNeil, L.K.; Zhu, D.; Perez, J.; Eiden, J.; Jansen, K.U.; et al. The discovery and development of a novel vaccine to protect against Neisseria meningitidis serogroup B disease. Hum. Vaccines Immunother. 2015, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Serruto, D.; Bottomley, M.J.; Ram, S.; Giuliani, M.M.; Rappuoli, R. The new multicomponent vaccine against meningococcal serogroup B, 4CMenB: Immunological, functional and structural characterization of the antigens. Vaccine 2012, 30 (Suppl. 2), B87–B97. [Google Scholar] [CrossRef] [PubMed]
- Łyskowski, A.; Leo, J.C.; Goldman, A. Structure and Biology of Trimeric Autotransporter Adhesins; Springer: Dordrecht, The Netherlands, 2011; Volume 715, pp. 143–158. [Google Scholar]
- Capecchi, B.; Adu-Bobie, J.; di Marcello, F.; Ciucchi, L.; Masignani, V.; Taddei, A.; Rappuoli, R.; Pizza, M.; Aricò, B. Neisseria meningitidis NadA is a new invasin which promotes bacterial adhesion to and penetration into human epithelial cells. Mol. Microbiol. 2005, 55, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Comanducci, M.; Bambini, S.; Brunelli, B.; Adu-Bobie, J.; Aricò, B.; Capecchi, B.; Giuliani, M.M.; Masignani, V.; Santini, L.; Savino, S.; et al. NadA, a novel vaccine candidate of Neisseria meningitidis. J. Exp. Med. 2002, 195, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Findlow, J.; Borrow, R.; Snape, M.D.; Dawson, T.; Holland, A.; John, T.M.; Evans, A.; Telford, K.L.; Ypma, E.; Toneatto, D.; et al. Multicenter, open-label, randomized phase II controlled trial of an investigational recombinant meningococcal serogroup B vaccine with and without outer membrane vesicles, administered in infancy. Clin. Infect. Dis. 2010, 51, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Bambini, S.; de Chiara, M.; Muzzi, A.; Mora, M.; Lucidarme, J.; Brehony, C.; Borrow, R.; Masignani, V.; Comanducci, M.; Maiden, M.C.J.; et al. Neisseria adhesin a variation and revised nomenclature scheme. Clin. Vaccine Immunol. CVI 2014, 21, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Dauter, Z. Solving coiled-coil protein structures. IUCrJ 2015, 2, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Dupeux, F.; Rower, M.; Seroul, G.; Blot, D.; Marquez, J.A. A thermal stability assay can help to estimate the crystallization likelihood of biological samples. Acta Crystallogr. D 2011, 67, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Malito, E.; Biancucci, M.; Faleri, A.; Ferlenghi, I.; Scarselli, M.; Maruggi, G.; Lo Surdo, P.; Veggi, D.; Liguori, A.; Santini, L.; et al. Structure of the meningococcal vaccine antigen NadA and epitope mapping of a bactericidal antibody. Proc. Natl. Acad. Sci. USA 2014, 111, 17128–17133. [Google Scholar] [CrossRef] [PubMed]
- Dale, G.E.; Oefner, C.; D’Arcy, A. The protein as a variable in protein crystallization. J. Struct. Biol. 2003, 142, 88–97. [Google Scholar] [CrossRef]
- Keenan, R.J.; Siehl, D.L.; Gorton, R.; Castle, L.A. DNA shuffling as a tool for protein crystallization. Proc. Natl. Acad. Sci. USA 2005, 102, 8887–8892. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.D.; Ridderbusch, O.; Zeth, K.; Albrecht, R.; Testa, O.; Woolfson, D.N.; Sauer, G.; Dunin-Horkawicz, S.; Lupas, A.N.; Alvarez, B.H. A coiled-coil motif that sequesters ions to the hydrophobic core. Proc. Natl. Acad. Sci. USA 2009, 106, 16950–16955. [Google Scholar] [CrossRef] [PubMed]
- Tavano, R.; Capecchi, B.; Montanari, P.; Franzoso, S.; Marin, O.; Sztukowska, M.; Cecchini, P.; Segat, D.; Scarselli, M.; Aricò, B.; et al. Mapping of the Neisseria meningitidis NadA cell-binding site: Relevance of predicted α-helices in the NH2-terminal and dimeric coiled-coil regions. J. Bacteriol. 2011, 193, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, R.P.; Smits, S.H.; Schmitt, L.; Slotboom, D.J.; Poolman, B. A structural classification of substrate-binding proteins. FEBS Lett. 2010, 584, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, F.; Fontana, M.R.; Soldaini, E.; Mishra, R.P.; Fiaschi, L.; Cartocci, E.; Nardi-Dei, V.; Ruggiero, P.; Nosari, S.; de Falco, M.G.; et al. Vaccine composition formulated with a novel TLR7-dependent adjuvant induces high and broad protection against Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2015, 112, 3680–3685. [Google Scholar] [PubMed]
- Mishra, R.P.; Mariotti, P.; Fiaschi, L.; Nosari, S.; Maccari, S.; Liberatori, S.; Fontana, M.R.; Pezzicoli, A.; de Falco, M.G.; Falugi, F.; et al. Staphylococcus aureus FhuD2 is involved in the early phase of staphylococcal dissemination and generates protective immunity in mice. J. Infect. Dis. 2012, 206, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, P.; Malito, E.; Biancucci, M.; Lo Surdo, P.; Mishra, R.P.; Nardi-Dei, V.; Savino, S.; Nissum, M.; Spraggon, G.; Grandi, G.; et al. Structural and functional characterization of the Staphylococcus aureus virulence factor and vaccine candidate FhuD2. Biochem. J. 2013, 449, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Podkowa, K.J.; Briere, L.A.; Heinrichs, D.E.; Shilton, B.H. Crystal and solution structure analysis of FhuD2 from Staphylococcus aureus in multiple unliganded conformations and bound to ferrioxamine-B. Biochemistry 2014, 53, 2017–2031. [Google Scholar] [CrossRef] [PubMed]
- Scully, I.L.; Liberator, P.A.; Jansen, K.U.; Anderson, A.S. Covering all the bases: Preclinical development of an effective Staphylococcus aureus vaccine. Front. Immunol. 2014, 5, 109. [Google Scholar] [CrossRef] [PubMed]
- Abate, F.; Malito, E.; Cozzi, R.; Lo Surdo, P.; Maione, D.; Bottomley, M.J. Apo, Zn2+-bound and Mn2+-bound structures reveal ligand-binding properties of SitA from the pathogen Staphylococcus pseudintermedius. Biosci. Rep. 2014, 34, e00154. [Google Scholar] [CrossRef] [PubMed]
- Gribenko, A.; Mosyak, L.; Ghosh, S.; Parris, K.; Svenson, K.; Moran, J.; Chu, L.; Li, S.; Liu, T.; Woods, V.L., Jr.; et al. Three-dimensional structure and biophysical characterization of Staphylococcus aureus cell surface antigen-manganese transporter MntC. J. Mol. Biol. 2013, 425, 3429–3445. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.S.; Scully, I.L.; Timofeyeva, Y.; Murphy, E.; McNeil, L.K.; Mininni, T.; Nunez, L.; Carriere, M.; Singer, C.; Dilts, D.A.; et al. Staphylococcus aureus manganese transport protein C is a highly conserved cell surface protein that elicits protective immunity against S. aureus and Staphylococcus epidermidis. J. Infect. Dis. 2012, 205, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, S.; Rouge, L.; Swem, D.L.; Sudhamsu, J.; Wu, P.; Russell, S.J.; Alexander, M.K.; Tam, C.; Nishiyama, M.; Starovasnik, M.A.; et al. Structural analysis of bacterial ABC transporter inhibition by an antibody fragment. Structure 2015, 23, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 2010, 17, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, G.; Ippolito, G.C.; Beausang, J.; Busse, C.E.; Wardemann, H.; Quake, S.R. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat. Biotechnol. 2014, 32, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Doria-Rose, N.A.; Longo, N.S.; Laub, L.; Lin, C.L.; Turk, E.; Kang, B.H.; Migueles, S.A.; Bailer, R.T.; Mascola, J.R.; et al. Isolation of human monoclonal antibodies from peripheral blood B cells. Nat. Protoc. 2013, 8, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.C.; Andrews, S.F. Tools to therapeutically harness the human antibody response. Nat. Rev. Immunol. 2012, 12, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, D.; Bottomley, M.J.; Malito, E.; Settembre, E.; Ferlenghi, I.; Cozzi, R. Advanced Vaccine Research; Bagnoli, F., Rappuoli, R., Eds.; Caister Academic Press: Norfolk, UK, 2015; pp. 103–132. [Google Scholar]
- Malito, E.; Faleri, A.; Lo Surdo, P.; Veggi, D.; Maruggi, G.; Grassi, E.; Cartocci, E.; Bertoldi, I.; Genovese, A.; Santini, L.; et al. Defining a protective epitope on factor H binding protein, a key meningococcal virulence factor and vaccine antigen. Proc. Natl. Acad. Sci. USA 2013, 110, 3304–3309. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.C.; Prosser, B.E.; Caesar, J.J.; Kugelberg, E.; Li, S.; Zhang, Q.; Quoraishi, S.; Lovett, J.E.; Deane, J.E.; Sim, R.B.; et al. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 2009, 458, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Giuntini, S.; Reason, D.C.; Granoff, D.M. Complement-mediated bactericidal activity of anti-factor H binding protein monoclonal antibodies against the meningococcus relies upon blocking factor H binding. Infect. Immun. 2011, 79, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gH–gL. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, C.; Chandramouli, S.; Donnarumma, D.; Nikitin, P.A.; Cianfrocco, M.A.; Gerrein, R.; Feire, A.L.; Barnett, S.W.; Lilja, A.E.; Rappuoli, R.; et al. Structural and biochemical studies of HCMV gH/gL/gO and pentamer reveal mutually exclusive cell entry complexes. Proc. Natl. Acad. Sci. USA 2015, 112, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Kringelum, J.V.; Nielsen, M.; Padkjaer, S.B.; Lund, O. Structural analysis of B-cell epitopes in antibody: Protein complexes. Mol. Immunol. 2013, 53, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Griffin, L.; Lawson, A. Antibody fragments as tools in crystallography. Clin. Exp. Immunol. 2011, 165, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Madico, G.; Welsch, J.A.; Lewis, L.A.; McNaughton, A.; Perlman, D.H.; Costello, C.E.; Ngampasutadol, J.; Vogel, U.; Granoff, D.M.; Ram, S. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J. Immunol. 2006, 177, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.C.; Exley, R.M.; Chan, H.; Feavers, I.; Kang, Y.H.; Sim, R.B.; Tang, C.M. Functional significance of factor H binding to Neisseria meningitidis. J. Immunol. 2006, 176, 7566–7575. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, M.M.; Biolchi, A.; Serruto, D.; Ferlicca, F.; Vienken, K.; Oster, P.; Rappuoli, R.; Pizza, M.; Donnelly, J. Measuring antigen-specific bactericidal responses to a multicomponent vaccine against serogroup B meningococcus. Vaccine 2010, 28, 5023–5030. [Google Scholar] [CrossRef] [PubMed]
- Masignani, V.; Comanducci, M.; Giuliani, M.M.; Bambini, S.; Adu-Bobie, J.; Arico, B.; Brunelli, B.; Pieri, A.; Santini, L.; Savino, S.; et al. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J. Exp. Med. 2003, 197, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Seib, K.L.; Serruto, D.; Oriente, F.; Delany, I.; Adu-Bobie, J.; Veggi, D.; Arico, B.; Rappuoli, R.; Pizza, M. Factor H-binding protein is important for meningococcal survival in human whole blood and serum and in the presence of the antimicrobial peptide LL-37. Infect. Immun. 2009, 77, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Cantini, F.; Veggi, D.; Dragonetti, S.; Savino, S.; Scarselli, M.; Romagnoli, G.; Pizza, M.; Banci, L.; Rappuoli, R. Solution structure of the factor H-binding protein, a survival factor and protective antigen of Neisseria meningitidis. J. Biol. Chem. 2009, 284, 9022–9026. [Google Scholar] [CrossRef] [PubMed]
- Cendron, L.; Veggi, D.; Girardi, E.; Zanotti, G. Structure of the uncomplexed Neisseria meningitidis factor H-binding protein fHbp (rLP2086). Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Mascioni, A.; Bentley, B.E.; Camarda, R.; Dilts, D.A.; Fink, P.; Gusarova, V.; Hoiseth, S.K.; Jacob, J.; Lin, S.L.; Malakian, K.; et al. Structural basis for the immunogenic properties of the meningococcal vaccine candidate LP2086. J. Biol. Chem. 2009, 284, 8738–8746. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Tan, L.; van der Veen, S.; Caesar, J.; Goicoechea De Jorge, E.; Harding, R.J.; Bai, X.; Exley, R.M.; Ward, P.N.; Ruivo, N.; et al. Design and evaluation of meningococcal vaccines through structure-based modification of host and pathogen molecules. PLoS Pathog. 2012, 8, e1002981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarselli, M.; Arico, B.; Brunelli, B.; Savino, S.; di Marcello, F.; Palumbo, E.; Veggi, D.; Ciucchi, L.; Cartocci, E.; Bottomley, M.J.; et al. Rational design of a meningococcal antigen inducing broad protective immunity. Sci. Transl. Med. 2011, 3, 91ra62. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, R.; Scarselli, M.; Ferlenghi, I. Structural vaccinology: A three-dimensional view for vaccine development. Curr. Top. Med. Chem. 2013, 13, 2629–2637. [Google Scholar] [PubMed]
- Dormitzer, P.R.; Grandi, G.; Rappuoli, R. Structural vaccinology starts to deliver. Nat. Rev. Microbiol. 2012, 10, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Beernink, P.T.; Shaughnessy, J.; Ram, S.; Granoff, D.M. Impaired immunogenicity of a meningococcal factor H-binding protein vaccine engineered to eliminate factor h binding. Clin. Vaccine Immunol. 2010, 17, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Beernink, P.T.; Shaughnessy, J.; Braga, E.M.; Liu, Q.; Rice, P.A.; Ram, S.; Granoff, D.M. A meningococcal factor H binding protein mutant that eliminates factor H binding enhances protective antibody responses to vaccination. J. Immunol. 2011, 186, 3606–3614. [Google Scholar] [CrossRef] [PubMed]
- Beernink, P.T.; Shaughnessy, J.; Pajon, R.; Braga, E.M.; Ram, S.; Granoff, D.M. The effect of human factor H on immunogenicity of meningococcal native outer membrane vesicle vaccines with over-expressed factor H binding protein. PLoS Pathog. 2012, 8, e1002688. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, S.; Johnson, S.; Jongerius, I.; Malik, T.; Genovese, A.; Santini, L.; Staunton, D.; Ufret-Vincenty, R.L.; Pickering, M.; Lea, S.M.; et al. Non-functional variant 3 factor H binding proteins as meningococcal vaccine candidates. Infect. Immun. 2014, 82, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Calmettes, C.; Alcantara, J.; Yu, R.H.; Schryvers, A.B.; Moraes, T.F. The structural basis of transferrin sequestration by transferrin-binding protein B. Nat. Struct. Mol. Biol. 2012, 19, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Noinaj, N.; Buchanan, S.K.; Cornelissen, C.N. The transferrin-iron import system from pathogenic Neisseria species. Mol. Microbiol. 2012, 86, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Noinaj, N.; Easley, N.C.; Oke, M.; Mizuno, N.; Gumbart, J.; Boura, E.; Steere, A.N.; Zak, O.; Aisen, P.; Tajkhorshid, E.; et al. Structural basis for iron piracy by pathogenic Neisseria. Nature 2012, 483, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Frandoloso, R.; Martinez-Martinez, S.; Calmettes, C.; Fegan, J.; Costa, E.; Curran, D.; Yu, R.H.; Gutierrez-Martin, C.B.; Rodriguez-Ferri, E.F.; Moraes, T.F.; et al. Nonbinding site-directed mutants of transferrin binding protein B exhibit enhanced immunogenicity and protective capabilities. Infect. Immun. 2015, 83, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- The IMpact-RSV-Study-Group. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 1998, 102, 531–537. [Google Scholar]
- Guvenel, A.K.; Chiu, C.; Openshaw, P.J. Current concepts and progress in RSV vaccine development. Expert Rev. Vaccines 2014, 13, 333–344. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.; Villafana, T.; Stillman, E.; Esser, M.T. Respiratory syncytial virus protein structure, function and implications for subunit vaccine development. Future Virol. 2014, 9, 753–767. [Google Scholar] [CrossRef]
- Anderson, R.; Huang, Y.; Langley, J.M. Prospects for defined epitope vaccines for respiratory syncytial virus. Future Microbiol. 2010, 5, 585–602. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Ray, W.C.; Peeples, M.E. Structure and function of respiratory syncytial virus surface glycoproteins. Curr. Top. Microbiol. Immunol. 2013, 372, 83–104. [Google Scholar] [PubMed]
- Calder, L.J.; Gonzalez-Reyes, L.; Garcia-Barreno, B.; Wharton, S.A.; Skehel, J.J.; Wiley, D.C.; Melero, J.A. Electron microscopy of the human respiratory syncytial virus fusion protein and complexes that it forms with monoclonal antibodies. Virology 2000, 271, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Liljeroos, L.; Krzyzaniak, M.A.; Helenius, A.; Butcher, S.J. Architecture of respiratory syncytial virus revealed by electron cryotomography. Proc. Natl. Acad. Sci. USA 2013, 110, 11133–11138. [Google Scholar] [CrossRef] [PubMed]
- Begona Ruiz-Arguello, M.; Gonzalez-Reyes, L.; Calder, L.J.; Palomo, C.; Martin, D.; Saiz, M.J.; Garcia-Barreno, B.; Skehel, J.J.; Melero, J.A. Effect of proteolytic processing at two distinct sites on shape and aggregation of an anchorless fusion protein of human respiratory syncytial virus and fate of the intervening segment. Virology 2002, 298, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.A.; Settembre, E.C.; Shaw, C.A.; Dey, A.K.; Rappuoli, R.; Mandl, C.W.; Dormitzer, P.R.; Carfi, A. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc. Natl. Acad. Sci. USA 2011, 108, 9619–9624. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pfarr, D.S.; Johnson, S.; Brewah, Y.A.; Woods, R.M.; Patel, N.K.; White, W.I.; Young, J.F.; Kiener, P.A. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J. Mol. Biol. 2007, 368, 652–665. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Chang, J.S.; Yang, Y.; Kim, A.; Graham, B.S.; Kwong, P.D. Structure of a major antigenic site on the respiratory syncytial virus fusion glycoprotein in complex with neutralizing antibody 101F. J. Virol. 2010, 84, 12236–12244. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Kim, A.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structural basis of respiratory syncytial virus neutralization by motavizumab. Nat. Struct. Mol. Biol. 2010, 17, 248–250. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 2011, 85, 7788–7796. [Google Scholar] [CrossRef] [PubMed]
- Magro, M.; Mas, V.; Chappell, K.; Vazquez, M.; Cano, O.; Luque, D.; Terron, M.C.; Melero, J.A.; Palomo, C. Neutralizing antibodies against the preactive form of respiratory syncytial virus fusion protein offer unique possibilities for clinical intervention. Proc. Natl. Acad. Sci. USA 2012, 109, 3089–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Correia, B.E.; Chen, M.; Yang, Y.; Graham, B.S.; Schief, W.R.; Kwong, P.D. Design and characterization of epitope-scaffold immunogens that present the motavizumab epitope from respiratory syncytial virus. J. Mol. Biol. 2011, 409, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nature 2014, 507, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Koff, W.C.; Russell, N.D.; Walport, M.; Feinberg, M.B.; Shiver, J.W.; Karim, S.A.; Walker, B.D.; McGlynn, M.G.; Nweneka, C.V.; Nabel, G.J. Accelerating the development of a safe and effective HIV vaccine: HIV vaccine case study for the Decade of Vaccines. Vaccine 2013, 31 (Suppl. 2), B204–B208. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Wilson, I.A. Insights into the trimeric HIV-1 envelope glycoprotein structure. Trends Biochem. Sci. 2015, 40, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Gilbert, P.B.; McElrath, M.J.; Zolla-Pazner, S.; Tomaras, G.D.; Alam, S.M.; Evans, D.T.; Montefiori, D.C.; Karnasuta, C.; Sutthent, R.; et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 2012, 366, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- West, A.P., Jr.; Scharf, L.; Scheid, J.F.; Klein, F.; Bjorkman, P.J.; Nussenzweig, M.C. Structural insights on the role of antibodies in HIV-1 vaccine and therapy. Cell 2014, 156, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Mascola, J.R.; Nabel, G.J. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: The end of the beginning. Nat. Rev. Immunol. 2013, 13, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [PubMed]
- Correia, B.E.; Ban, Y.E.; Holmes, M.A.; Xu, H.; Ellingson, K.; Kraft, Z.; Carrico, C.; Boni, E.; Sather, D.N.; Zenobia, C.; et al. Computational design of epitope-scaffolds allows induction of antibodies specific for a poorly immunogenic HIV vaccine epitope. Structure 2010, 18, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, M.L.; Correia, B.E.; Ban, Y.-E.A.; Carrico, C.; Kalyuzhniy, O.; Chen, L.; Schroeter, A.; Huang, P.-S.; Mclellan, J.S.; Kwong, P.D.; et al. Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science 2011, 334, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Ofek, G.; Guenaga, F.J.; Schief, W.R.; Skinner, J.; Baker, D.; Wyatt, R.; Kwong, P.D. Elicitation of structure-specific antibodies by epitope scaffolds. Proc. Natl. Acad. Sci. USA 2010, 107, 17880–17887. [Google Scholar] [CrossRef] [PubMed]
- Dey, B.; Svehla, K.; Xu, L.; Wycuff, D.; Zhou, T.; Voss, G.; Phogat, A.; Chakrabarti, B.K.; Li, Y.; Shaw, G.; et al. Structure-based stabilization of HIV-1 gp120 enhances humoral immune responses to the induced co-receptor binding site. PLoS Pathog. 2009, 5, e1000445. [Google Scholar] [CrossRef] [PubMed]
- Kassa, A.; Dey, A.K.; Sarkar, P.; Labranche, C.; Go, E.P.; Clark, D.F.; Sun, Y.; Nandi, A.; Hartog, K.; Desaire, H.; et al. Stabilizing exposure of conserved epitopes by structure guided insertion of disulfide bond in HIV-1 envelope glycoprotein. PLoS ONE 2013, 8, e76139. [Google Scholar] [CrossRef] [PubMed]
- Jardine, J.; Julien, J.P.; Menis, S.; Ota, T.; Kalyuzhniy, O.; McGuire, A.; Sok, D.; Huang, P.S.; MacPherson, S.; Jones, M.; et al. Rational HIV immunogen design to target specific germline B cell receptors. Science 2013, 340, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.P.; Cupo, A.; Sok, D.; Stanfield, R.L.; Lyumkis, D.; Deller, M.C.; Klasse, P.J.; Burton, D.R.; Sanders, R.W.; Moore, J.P.; et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 2013, 342, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Pancera, M.; Zhou, T.; Druz, A.; Georgiev, I.S.; Soto, C.; Gorman, J.; Huang, J.; Acharya, P.; Chuang, G.Y.; Ofek, G.; et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 2014, 514, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lyumkis, D.; Julien, J.P.; de Val, N.; Cupo, A.; Potter, C.S.; Klasse, P.J.; Burton, D.R.; Sanders, R.W.; Moore, J.P.; Carragher, B.; et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 2013, 342, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Derking, R.; Cupo, A.; Julien, J.P.; Yasmeen, A.; de Val, N.; Kim, H.J.; Blattner, C.; de la Pena, A.T.; Korzun, J.; et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013, 9, e1003618. [Google Scholar] [CrossRef] [PubMed]
- Hol, W.; Verlinde, C. Macromolecular Crystallography and Medicine; Springer: Dordrecht, UK; Boston, UK; London, UK, 2006; Volume F, pp. 10–25. [Google Scholar]
- Tickle, I.; Sharff, A.; Vinkovic, M.; Yon, J.; Jhoti, H. High-throughput protein crystallography and drug discovery. Chem. Soc. Rev. 2004, 33, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Kleywegt, G.J.; Nakamura, H.; Markley, J.L. The protein data bank at 40: Reflecting on the past to prepare for the future. Structure (Lond., Engl.) 2012, 20, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.C.; Stuart, D.; Yokoyama, S. Lessons from structural genomics. Ann. Rev. Biophys. 2009, 38, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J.D.; Crowfoot, D. X-ray photographs of crystalline pepsin. Nature 1934, 133, 794–795. [Google Scholar] [CrossRef]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.G.; Wyckoff, H.; Phillips, D.C. A three-dimensional model of the myoglobin molecule obtained by X-ray analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Kendrew, J.C.; Dickerson, R.E.; Strandberg, B.E.; Hart, R.G.; Davies, D.R.; Phillips, D.C.; Shore, V.C. Structure of myoglobin: A three-dimensional Fourier synthesis at 2 Å. resolution. Nature 1960, 185, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F.; Rossmann, M.G.; Cullis, A.F.; Muirhead, H.; Will, G.; North, A.C. Structure of haemoglobin: A three-dimensional Fourier synthesis at 5.5-A. resolution, obtained by X-ray analysis. Nature 1960, 185, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.; Fenn, R.H.; North, A.C.; Phillips, D.C.; Poljak, R.J. Structure of lysozyme. A Fourier map of the electron density at 6 angstrom resolution obtained by X-ray diffraction. Nature 1962, 196, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.; Koenig, D.F.; Mair, G.A.; North, A.C.; Phillips, D.C.; Sarma, V.R. Structure of hen egg-white lysozyme. A three-dimensional Fourier synthesis at 2 Angstrom resolution. Nature 1965, 206, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Garman, E.F. Developments in X-ray crystallographic structure determination of biological macromolecules. Science 2014, 343, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Chayen, N.E.; Saridakis, E. Protein crystallization: From purified protein to diffraction-quality crystal. Nat. Methods 2008, 5, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, M.A.; Grütter, M.G. New concepts and aids to facilitate crystallization. Curr. Opin. Struct. Biol. 2013, 23, 409–416. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Cudney, B. Optimization of crystallization conditions for biological macromolecules. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 1445–1467. [Google Scholar] [CrossRef] [PubMed]
- Chandonia, J.-M.; Brenner, S.E. The impact of structural genomics: Expectations and outcomes. Science 2006, 311, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, W.A. Impact of structures from the protein structure initiative. Structure (Lond., Engl.) 2007, 15, 1528–1529. [Google Scholar] [CrossRef] [PubMed]
- Rupp, B. High-throughput crystallography at an affordable cost: The TB structural genomics consortium crystallization facility. Acc. Chem. Res. 2003, 36, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Arrowsmith, C.H.; Allali-Hassani, A.; Senisterra, G.; Wasney, G.A. Biophysical characterization of recombinant proteins: A key to higher structural genomics success. J. Struct. Biol. 2010, 172, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Yumerefendi, H.; Tarendeau, F.; Mas, P.J.; Hart, D.J. ESPRIT: An automated, library-based method for mapping and soluble expression of protein domains from challenging targets. J. Struct. Biol. 2010, 172, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.; Puckey, L.H.; Cheetham, C.L.; Harris, R.; Ali, A.A.; Bhattacharyya, U.; Maclagan, K.; Powell, K.A.; Prodromou, C.; Pearl, L.H.; et al. Combinatorial domain hunting: An effective approach for the identification of soluble protein domains adaptable to high-throughput applications. Protein Sci. 2006, 15, 2356–2365. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S. Application of protein engineering to enhance crystallizability and improve crystal properties. Acta Crystallogr. D 2010, 66, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Lamazares, E.; Clemente, I.; Bueno, M.; Velazquez-Campoy, A.; Sancho, J. Rational stabilization of complex proteins: A divide and combine approach. Sci. Rep. 2015, 5, 9129. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S.; Vekilov, P.G. Entropy and surface engineering in protein crystallization. Acta Crystallogr. D 2006, 62, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Weis, W.I.; Kobilka, B.K. N-terminal T4 lysozyme fusion facilitates crystallization of a G protein coupled receptor. PLoS ONE 2012, 7, e46039. [Google Scholar] [CrossRef] [PubMed]
- Chun, E.; Thompson, A.A.; Liu, W.; Roth, C.B.; Griffith, M.T.; Katritch, V.; Kunken, J.; Xu, F.; Cherezov, V.; Hanson, M.A.; et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 2012, 20, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Hassell, A.M.; An, G.; Bledsoe, R.K.; Bynum, J.M.; Carter, H.L.; Deng, S.-J.J.; Gampe, R.T.; Grisard, T.E.; Madauss, K.P.; Nolte, R.T.; et al. Crystallization of protein-ligand complexes. Acta Crystallogr. D 2007, 63, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Tereshko, V.; Uysal, S.; Koide, A.; Margalef, K.; Koide, S.; Kossiakoff, A.A. Toward chaperone-assisted crystallography: Protein engineering enhancement of crystal packing and X-ray phasing capabilities of a camelid single-domain antibody (VHH) scaffold. Protein Sci. Publ. Protein Soc. 2008, 17, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.; Edwards, A. In situ proteolysis to generate crystals for structure determination: An update. PLoS ONE 2009, 4, e5094. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, A.; Villard, F.; Marsh, M. An automated microseed matrix-screening method for protein crystallization. Acta crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Bergfors, T. Seeds to crystals. J. Struct. Biol. 2003, 142, 66–76. [Google Scholar] [CrossRef]
- Walter, T.S.; Meier, C.; Assenberg, R.; Au, K.-F.; Ren, J.; Verma, A.; Nettleship, J.E.; Owens, R.J.; Stuart, D.I.; Grimes, J.M. Lysine methylation as a routine rescue strategy for protein crystallization. Structure (Lond., Engl.) 2006, 14, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Quartey, P.; Li, H.; Volkart, L.; Hatzos, C.; Chang, C.; Nocek, B.; Cuff, M.; Osipiuk, J.; Tan, K.; et al. Large-scale evaluation of protein reductive methylation for improving protein crystallization. Nat. Methods 2008, 5, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.; Bolton, E.E.; Mueller-Dieckmann, J.; Fazio, V.J.; Gallagher, D.T.; Lovell, D.; Luft, J.R.; Peat, T.S.; Ratcliffe, D.; Sayle, R.A.; et al. On the need for an international effort to capture, share and use crystallization screening data. Acta Crystallogr. F 2012, 68, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Fazio, V.J.; Peat, T.S.; Newman, J. A drunken search in crystallization space. Acta Crystallogr. F 2014, 70, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Fusco, D.; Barnum, T.J.; Bruno, A.E.; Luft, J.R.; Snell, E.H.; Mukherjee, S.; Charbonneau, P. Statistical analysis of crystallization database links protein physico-chemical features with crystallization mechanisms. PLoS ONE 2014, 9, e101123. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.T.; Dekker, C.; Kroemer, M.; Osborne, M.; von Delft, F. Using textons to rank crystallization droplets by the likely presence of crystals. Acta Crystallogr. D 2014, 70, 2702–2718. [Google Scholar] [CrossRef] [PubMed]
- Dauter, Z.; Jaskolski, M.; Wlodawer, A. Impact of synchrotron radiation on macromolecular crystallography: A personal view. J. Synchrotron Radiat. 2010, 17, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Broennimann, C.; Eikenberry, E.F.; Henrich, B.; Horisberger, R.; Huelsen, G.; Pohl, E.; Schmitt, B.; Schulze-Briese, C.; Suzuki, M.; Tomizaki, T.; et al. The PILATUS 1M detector. J. Synchrotron Radiat. 2006, 13, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Wang, M.; Schulze-Briese, C. Optimal fine φ-slicing for single-photon-counting pixel detectors. Acta Crystallogr. D 2012, 68, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Gabadinho, J.; Beteva, A.; Guijarro, M.; Rey-Bakaikoa, V.; Spruce, D.; Bowler, M.W.; Brockhauser, S.; Flot, D.; Gordon, E.J.; Hall, D.R.; et al. MxCuBE: A synchrotron beamline control environment customized for macromolecular crystallography experiments. J. Synchrotron Radiat. 2010, 17, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, J.R.; Mitchell, E.P. Synchrotron radiation macromolecular crystallography: Science and spin-offs. IUCrJ 2015, 2, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Malbet-Monaco, S.; Leonard, G.A.; Mitchell, E.P.; Gordon, E.J. How the ESRF helps industry and how they help the ESRF. Acta Crystallogr. D 2013, 69, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, I. Serial femtosecond crystallography: The first five years. IUCrJ 2015, 2, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L.; et al. High-resolution protein structure determination by serial femtosecond crystallography. Science 2012, 337, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wacker, D.; Gati, C.; Han, G.W.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; Katritch, V.; Barty, A.; et al. Serial femtosecond crystallography of G protein-coupled receptors. Science 2013, 342, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Tenboer, J.; Basu, S.; Zatsepin, N.; Pande, K.; Milathianaki, D.; Frank, M.; Hunter, M.; Boutet, S.; Williams, G.J.; Koglin, J.E.; et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 2014, 346, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, W.A. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science 1991, 254, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Garman, E.; Murray, J.W. Heavy-atom derivatization. Acta Crystallogr. D 2003, 59, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Rould, M.A. The same but different: Isomorphous methods for phasing and high-throughput ligand screening. Methods Mol. Biol. 2007, 364, 159–182. [Google Scholar] [PubMed]
- Gonzalez, A.; von Delft, F.; Liddington, R.C.; Bakolitsa, C. Two-wavelength MAD phasing and radiation damage: A case study. J. Synchrotron Radiat. 2005, 12, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Dodson, E. Is it jolly SAD? Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 1958–1965. [Google Scholar] [CrossRef]
- Dauter, Z. One-and-a-half wavelength approach. Acta Crystallogr. D 2002, 58, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Abendroth, J.; Gardberg, A.S.; Robinson, J.I.; Christensen, J.S.; Staker, B.L.; Myler, P.J.; Stewart, L.J.; Edwards, T.E. SAD phasing using iodide ions in a high-throughput structural genomics environment. J. Struct. Funct. Genomics 2011, 12, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Dauter, Z. New approaches to high-throughput phasing. Curr. Opin. Struct. Biol. 2002, 12, 674–678. [Google Scholar] [CrossRef]
- Dauter, Z.; Li, M.; Wlodawer, A. Practical experience with the use of halides for phasing macromolecular structures: A powerful tool for structural genomics. Acta Crystallogr. D 2001, 57, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Nagem, R.A.; Dauter, Z.; Polikarpov, I. Protein crystal structure solution by fast incorporation of negatively and positively charged anomalous scatterers. Acta Crystallogr. D 2001, 57, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Scapin, G. Molecular replacement then and now. Acta Crystallogr. D 2013, 69, 2266–2275. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.; McCoy, A. An introduction to molecular replacement. Acta Crystallogr. D 2008, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abergel, C. Molecular replacement: Tricks and treats. Acta Crystallogr. D 2013, 69, 2167–2173. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Long, F.; Vagin, A.A.; Young, P.; Murshudov, G.N. BALBES: A molecular-replacement pipeline. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Keegan, R.M.; Winn, M.D. MrBUMP: An automated pipeline for molecular replacement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Stokes-Rees, I.; Sliz, P. Protein structure determination by exhaustive search of Protein Data Bank derived databases. Proc. Natl. Acad. Sci. USA 2010, 107, 21476–21481. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Morin, A.; Eisenbraun, B.; Key, J.; Sanschagrin, P.C.; Timony, M.A.; Ottaviano, M.; Sliz, P. Collaboration gets the most out of software. eLife 2013, 2, e01456. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.T.; Settembre, E.C.; Trask, S.D.; Greenberg, H.B.; Harrison, S.C.; Dormitzer, P.R. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 2009, 324, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Mooney, P.; Zheng, S.; Booth, C.R.; Braunfeld, M.B.; Gubbens, S.; Agard, D.A.; Cheng, Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 2013, 10, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Settembre, E.; Xu, C.; Dormitzer, P.R.; Bellamy, R.; Harrison, S.C.; Grigorieff, N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc. Natl. Acad. Sci. USA 2008, 105, 1867–1872. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Amunts, A.; Bai, X.C.; Sugimoto, Y.; Edwards, P.C.; Murshudov, G.; Scheres, S.H.; Ramakrishnan, V. Structure of the large ribosomal subunit from human mitochondria. Science 2014, 346, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Avila-Sakar, A.; Kim, J.; Booth, D.S.; Greenberg, C.H.; Rossi, A.; Liao, M.; Li, X.; Alian, A.; Griner, S.L.; et al. Fabs enable single particle cryoEM studies of small proteins. Structure 2012, 20, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Noble, K.A.; Mao, Y.; Young, N.L.; Sathe, S.K.; Roux, K.H.; Marshall, A.G. Rapid screening for potential epitopes reactive with a polycolonal antibody by solution-phase H/D exchange monitored by FT-ICR mass spectrometry. J. Am. Soc. Mass Spectrom. 2013, 24, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, R.R.; Chalmers, M.J.; Griffin, P.R. Automated hydrogen/deuterium exchange electron transfer dissociation high resolution mass spectrometry measured at single-amide resolution. J. Am. Soc. Mass Spectrom. 2012, 23, 301–309. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malito, E.; Carfi, A.; Bottomley, M.J. Protein Crystallography in Vaccine Research and Development. Int. J. Mol. Sci. 2015, 16, 13106-13140. https://doi.org/10.3390/ijms160613106
Malito E, Carfi A, Bottomley MJ. Protein Crystallography in Vaccine Research and Development. International Journal of Molecular Sciences. 2015; 16(6):13106-13140. https://doi.org/10.3390/ijms160613106
Chicago/Turabian StyleMalito, Enrico, Andrea Carfi, and Matthew J. Bottomley. 2015. "Protein Crystallography in Vaccine Research and Development" International Journal of Molecular Sciences 16, no. 6: 13106-13140. https://doi.org/10.3390/ijms160613106
APA StyleMalito, E., Carfi, A., & Bottomley, M. J. (2015). Protein Crystallography in Vaccine Research and Development. International Journal of Molecular Sciences, 16(6), 13106-13140. https://doi.org/10.3390/ijms160613106