
A Mechanism-Based Model for the Prediction of the Metabolic Sites of Steroids Mediated by Cytochrome P450 3A4


Abstract
:1. Introduction
2. Results and Discussion
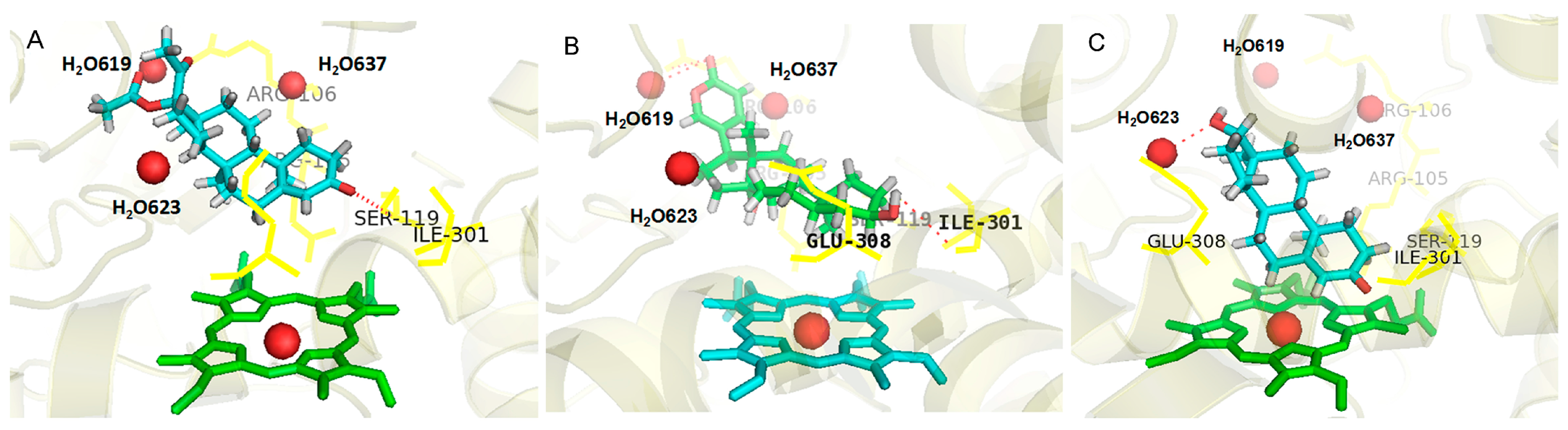
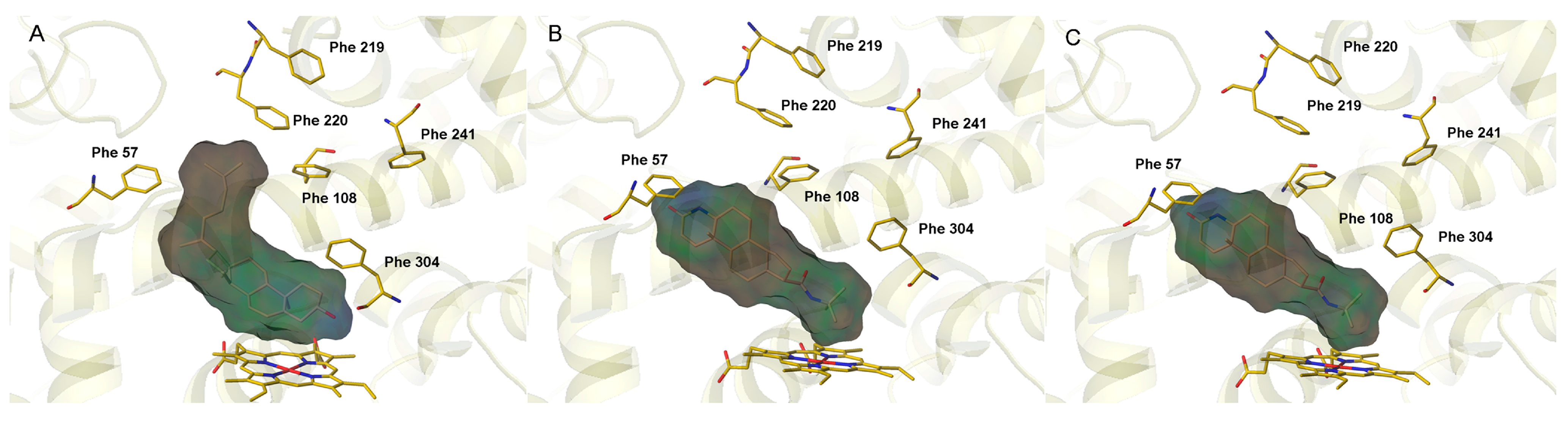
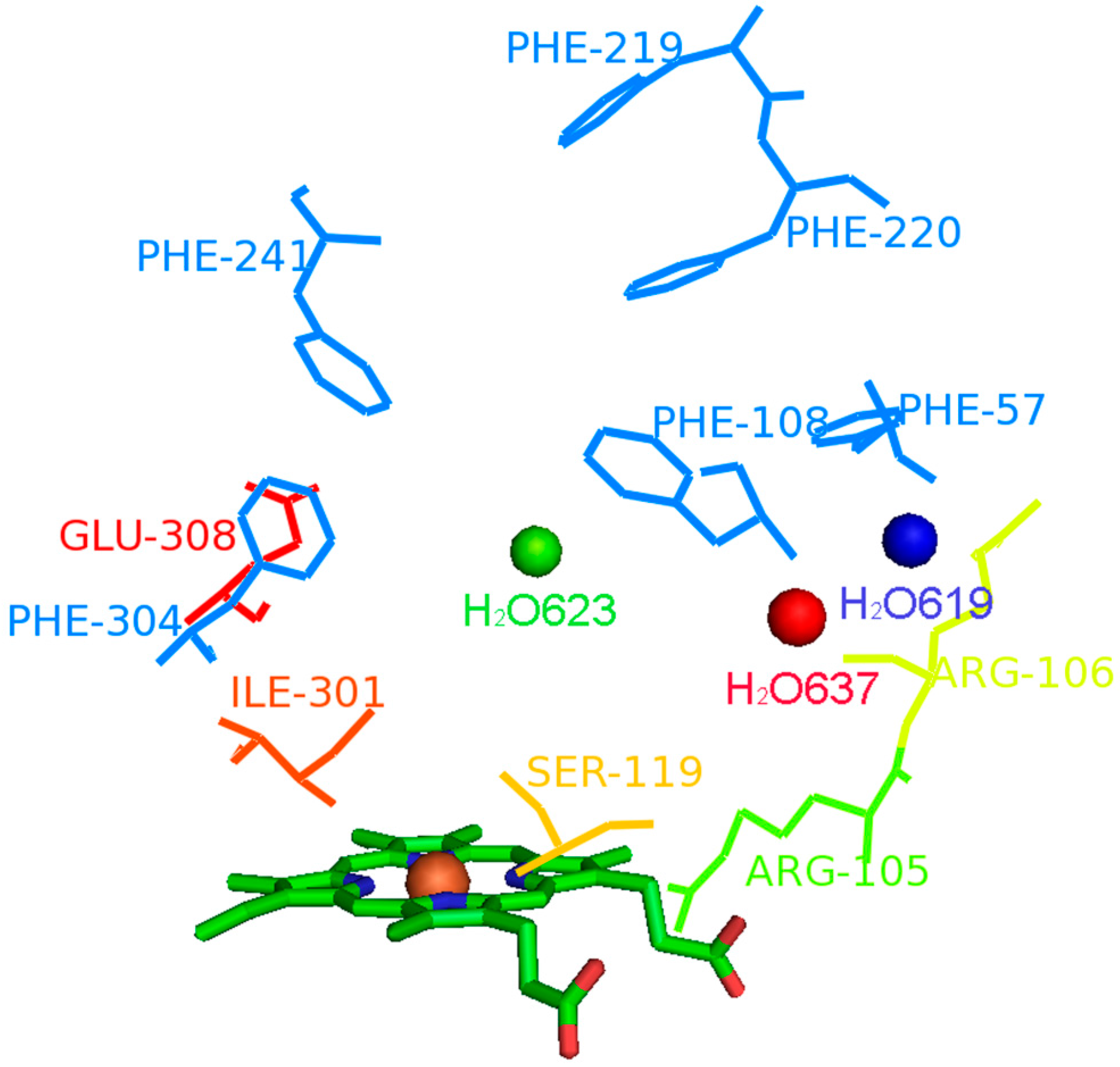
2.1. Analysis of Active Conformations


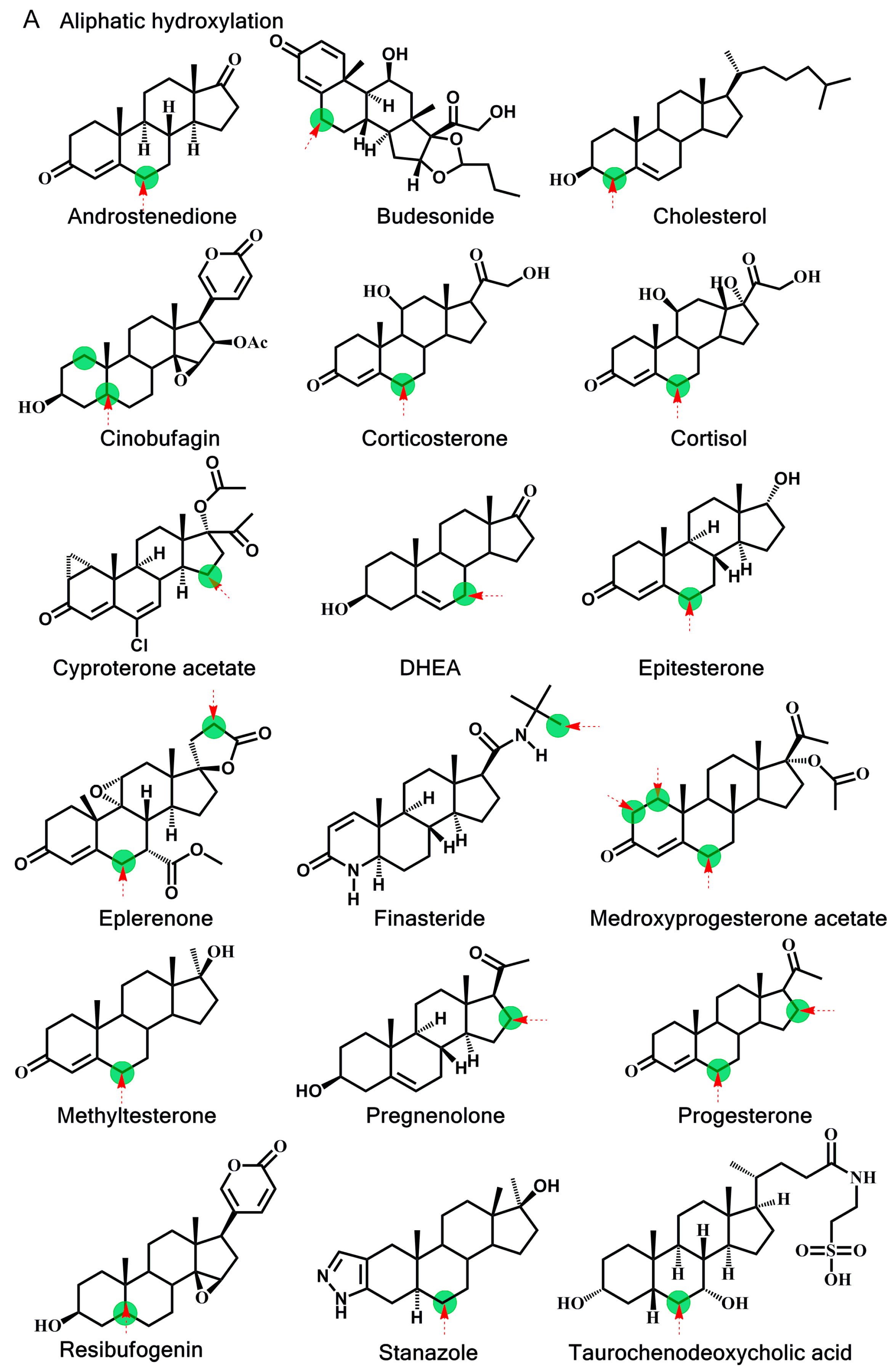
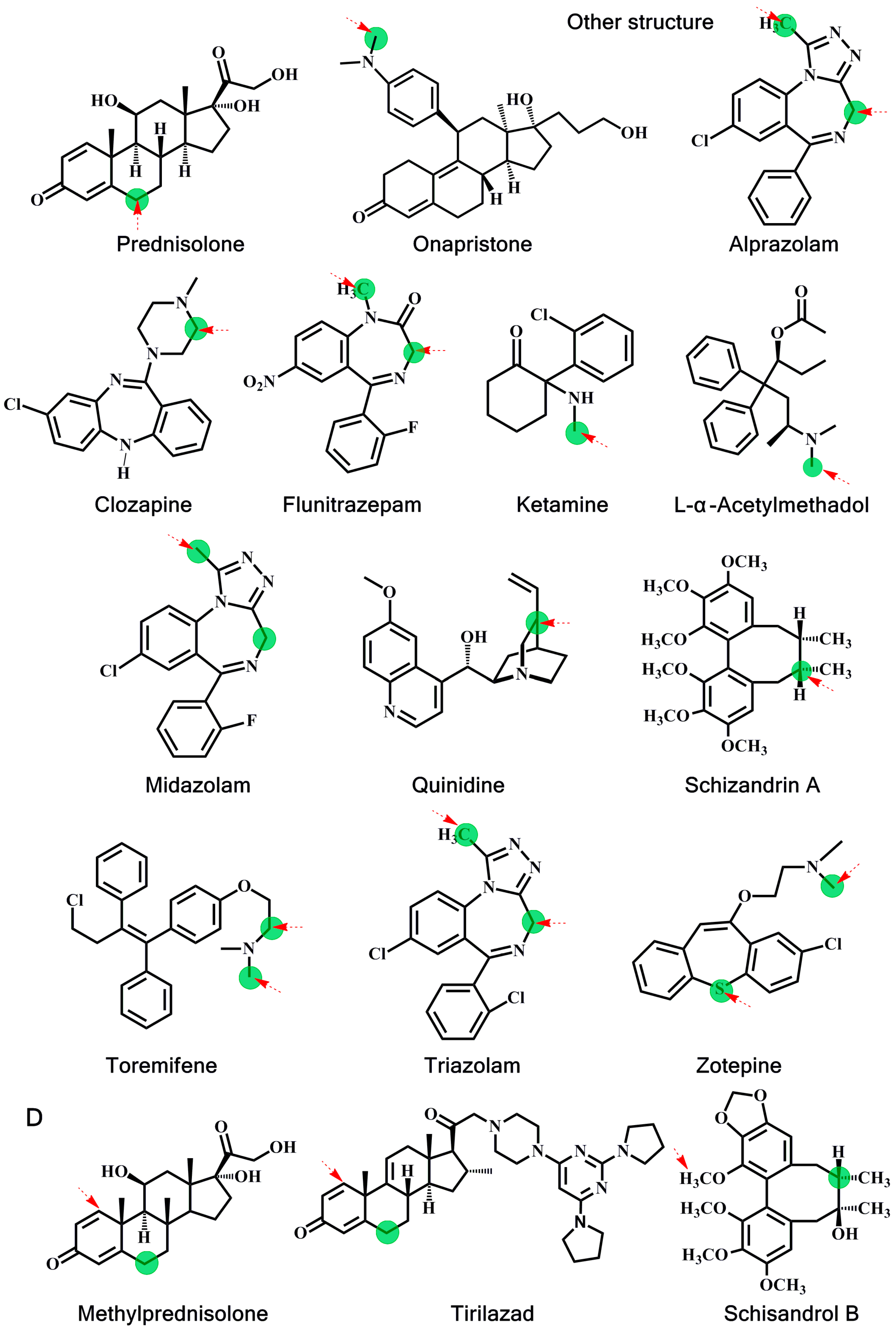
2.2. Estimation of Activation Energy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Types | Model of Activation Energy Estimation | ||
|---|---|---|---|
| Nc a | N b | Ratio (%) c | |
| Aliphatic hydroxylation | 23 | 40 | 57.50% |
| N-dealkylation | 8 | 10 | 80.00% |
| Total | 31 | 50 | 62.00% |
2.3. Application of the Mechanism-Based Model
| Reaction Types | Training Set | ||
|---|---|---|---|
| Nc a | N b | Ratio (%) c | |
| Aliphatic hydroxylation | 20 | 25 | 80.00% |
| N-dealkylation | 3 | 3 | 100% |
| Total | 23 | 28 | 82.14% |
| Reaction Types | Test Set | ||
|---|---|---|---|
| Nc b | N a | Ratio (%) c | |
| Aliphatic hydroxylation | 12 | 15 | 80.00% |
| N-dealkylation | 7 | 7 | 100% |
| Total | 19 | 22 | 86.36% |
2.4. Analysis of the Prediction Model
3. Experimental Section
3.1. Dataset and Molecule Modeling



3.2. Generating Active Conformations
3.3. Activation Energy Estimation
3.4. Prediction Models


4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brown, D. Unfinished business: Target-based drug discovery. Drug Discov. Today 2007, 12, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Nakasa, H.; Ishii, I.; Ariyoshi, N.; Igarashi, T.; Ohmori, S.; Kitada, M. Effects of endogenous steroids on CYP3A4-mediated drug metabolism by human liver microsomes. Drug Metab. Dispos.: Biol. Fate Chem. 2002, 30, 534–540. [Google Scholar] [CrossRef]
- Zaretzki, J.; Bergeron, C.; Rydberg, P.; Huang, T.W.; Bennett, K.P.; Breneman, C.M. RS-predictor: A new tool for predicting sites of cytochrome P450-mediated metabolism applied to CYP 3A4. J. Chem. Inf. Model. 2011, 51, 1667–1689. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Gloriam, D.E.; Zaretzki, J.; Breneman, C.; Olsen, L. SMARTCyp: A 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Med. Chem. Lett. 2010, 1, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Blair, I.A. Analysis of estrogens in serum and plasma from postmenopausal women: Past present, and future. Steroids 2010, 75, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.V.; Ogg, M.S.; Goldfarb, P.S.; Gibson, G.G. Molecular modelling of the human glucocorticoid receptor (hGR) ligand-binding domain (LBD) by homology with the human estrogen receptor alpha (hER α) LBD: Quantitative structure-activity relationships within a series of CYP3A4 inducers where induction is mediated via hGR involvement. J. Steroid Biochem. 2002, 82, 195–199. [Google Scholar]
- Yamazaki, H.; Shimada, T. Progesterone and testosterone hydroxylation by cytochromes P450 2C19, 2C9, and 3A4 in human liver microsomes. Arch. Biochem. Biophys. 1997, 346, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Nolteernsting, E.; Schanzer, W. Metabolism of anabolic steroids by recombinant human cytochrome P450 enzymes. Gas chromatographic-mass spectrometric determination of metabolites. J. Chromatogr. B 1999, 735, 73–83. [Google Scholar] [CrossRef]
- Wang, Y.H.; Han, K.L.; Yang, S.L.; Yang, L. Structural determinants of steroids for cytochrome P450 3A4-mediated metabolism. J. Mol. Struct. 2004, 710, 215–221. [Google Scholar] [CrossRef]
- Zhang, J.W.; Liu, Y.; Zhao, J.Y.; Wang, L.M.; Ge, G.B.; Gao, Y.; Li, W.; Liu, H.T.; Liu, H.X.; Zhang, Y.Y.; et al. Metabolic profiling and cytochrome P450 reaction phenotyping of medroxyprogesterone acetate. Drug Metab. Dispos. 2008, 36, 2292–2228. [Google Scholar] [CrossRef] [PubMed]
- Siemes, C.; Visser, L.E.; de Jong, F.H.; Coebergh, J.W.W.; Uitterlinden, A.G.; Hofman, A.; Stricker, B.H.C.; van Schaik, R.H.N. Cytochrome P450 3A gene variation, steroid hormone serum levels and prostate cancer—The Rotterdam Study. Steroids 2010, 75, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.C.; Hines, R.N.; Gu, C.; Koukouritaki, S.B.; Manro, J.R.; Tandler, P.J.; Zaya, M.J. Developmental expression of the major human hepatic CYP3A enzymes. J. Pharmacol. Exp. Ther. 2003, 307, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Luu-The, V. Assessment of steroidogenesis and steroidogenic enzyme functions. J. Steroid Biochem. Mol. Biol. 2013, 137, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metab. Rev. 2002, 34, 83–448. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.P.; Potter, B.V. The structural biology of oestrogen metabolism. J. Steroid Biochem. Mol. Biol. 2013, 137, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Stjernschantz, E.; Vermeulen, N.P.; Oostenbrink, C. Computational prediction of drug binding and rationalisation of selectivity towards cytochromes P450. Expert Opin. Drug Metab. Toxicol. 2008, 4, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Klopman, G.; Dimayuga, M.; Talafous, J. META. 1. A program for the evaluation of metabolic transformation of chemicals. J. Chem. Inf. Comput. Sci. 1994, 34, 1320–1325. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.C.; Hawksworth, G.; Horne, V.A.; Newlands, A.; Morsman, J.; Tute, M.S.; Smith, D.A. Putative active site template model for cytochrome P4502C9 (tolbutamide hydroxylase). Drug Metab. Dispos. 1996, 24, 260–266. [Google Scholar] [PubMed]
- Borodina, Y.; Rudik, A.; Filimonov, D.; Kharchevnikova, N.; Dmitriev, A.; Blinova, V.; Poroikov, V. A new statistical approach to predicting aromatic hydroxylation sites. Comparison with model-based approaches. J. Chem. Inf. Comput. Sci. 2004, 44, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Shen, L.Q.; Walker, M.J.; Sheridan, R.P. A model for predicting likely sites of CYP3A4-mediated metabolism on drug-like molecules. J. Med. Chem. 2003, 46, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Moors, S.L.; Vos, A.M.; Cummings, M.D.; van Vlijmen, H.; Ceulemans, A. Structure-based site of metabolism prediction for cytochrome P450 2D6. J. Med. Chem. 2011, 54, 6098–6105. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, J.; Tawa, G.; Wallqvist, A. 2D SMARTCyp reactivity-based site of metabolism prediction for major drug-metabolizing cytochrome P450 enzymes. J. Chem. Inf. Model. 2012, 52, 1698–1712. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, R.P.; Korzekwa, K.R.; Torres, R.A.; Walker, M.J. Empirical regioselectivity models for human cytochromes P450 3A4, 2D6, and 2C9. J. Med. Chem. 2007, 50, 3173–3184. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Williamson, M.J.; Tyzack, J.D.; Tan, L.; Bond, P.J.; Bender, A.; Glen, R.C. Computational prediction of metabolism: Sites, products, SAR, P450 enzyme dynamics, and mechanisms. J. Chem. Inf. Model. 2012, 52, 617–648. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, N.D.; Kim, S.Y.; Choi, I.; Cho, K.H.; Oh, W.S.; Kim, D.N.; No, K.T. Regioselectivity prediction of CYP1A2-mediated phase I metabolism. J. Chem. Inf. Model. 2008, 48, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, C.; Vermeulen, N.P.E.; Feenstra, K.A. Cytochrome P450 in silico: An integrative modeling approach. J. Med. Chem. 2005, 48, 2725–2755. [Google Scholar]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 2002, 124, 11809–11826. [Google Scholar]
- Olsen, L.; Rydberg, P.; Rod, T.H.; Ryde, U. Prediction of activation energies for hydrogen abstraction by cytochrome P450. J. Med. Chem. 2006, 49, 6489–6499. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Krauser, J.A.; Johnson, W.W. Rate-limiting steps in oxidations catalyzed by rabbit cytochrome P450 1A2. Biochemistry 2004, 43, 10775–10788. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Poulos, T.L. Interaction of human cytochrome P4503A4 with ritonavir analogs. Arch. Biochem. Biophys. 2012, 520, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Wu, B.; Chow, D.; Hu, M. Substrate selectivity of drug-metabolizing cytochrome P450s predicted from crystal structures and in silico modeling. Drug Metab. Rev. 2012, 44, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Vasanthanathan, P.; Hritz, J.; Taboureau, O.; Olsen, L.; Jorgensen, F.S.; Vermeulen, N.P.; Oostenbrink, C. Virtual screening and prediction of site of metabolism for cytochrome P450 1A2 ligands. J. Chem. Inf. Model. 2009, 49, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Korolev, D.; Balakin, K.V.; Nikolsky, Y.; Kirillov, E.; Ivanenkov, Y.A.; Savchuk, N.P.; Ivashchenko, A.A.; Nikolskaya, T. Modeling of human cytochrome P450-mediated drug metabolism using unsupervised machine learning approach. J. Med. Chem. 2003, 46, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2887. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; McClusky, G.A. Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. Biochem. Biophys. Res. Commun. 1978, 81, 154–160. [Google Scholar] [CrossRef]
- Okazaki, O.; Guengerich, F.P. Evidence for specific base catalysis in N-dealkylation reactions catalyzed by cytochrome P450 and chloroperoxidase. Differences in rates of deprotonation of aminium radicals as an explanation for high kinetic hydrogen isotope effects observed with peroxidases. J. Biol. Chem. 1993, 268, 1546–1552. [Google Scholar] [PubMed]
- Li, D.M.; Wang, Y.; Yang, C.L.; Han, K.L. Theoretical study of N-dealkylation of N-cyclopropyl-N-methylaniline catalyzed by cytochrome P450: Insight into the origin of the regioselectivity. Dalton Trans. 2009, 14, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.Z. A literature review of enzyme kinetic parameters for CYP3A4-mediated metabolic reactions of 113 drugs in human liver microsomes: Structure-kinetics relationship assessment. Curr. Drug Metab. 2006, 7, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Yu, Z.L.; Hu, L.H.; Wang, C.; Huo, X.K.; Deng, S.; Hou, J.; Wu, J.J.; Ge, G.B.; Ma, X.C.; et al. Characterization of phase I metabolism of resibufogenin and evaluation of the metabolic effects on its antitumor activity and toxicity. Drug Metab. Dispos. 2015, 43, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Ge, G.B.; Ning, J.; Hu, L.H.; Dai, Z.R.; Hou, J.; Cao, Y.F.; Yu, Z.W.; Ai, C.Z.; Gu, J.K.; Ma, X.C.; et al. A highly selective probe for human cytochrome P450 3A4: isoform selectivity, kinetic characterization and its applications. Chem. Commun. 2013, 49, 9779–9781. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.C.; Ning, J.; Ge, G.B.; Liang, S.C.; Wang, X.L.; Zhang, B.J.; Huang, S.S.; Li, J.K.; Yang, L. Comparative metabolism of cinobufagin in liver microsomes from mouse, rat, dog, minipig, monkey, and human. Drug Metab. Dispos. 2011, 39, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A.; Dobler, M.; Vedani, A. Prediction of small-molecule binding to cytochrome P450 3A4: Flexible docking combined with multidimensional QSAR. Chemmedchem 2006, 1, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Narasimhulu, S. Differential behavior of the sub-sites of cytochrome 450 active site in binding of substrates, and products (implications for coupling/uncoupling). Biochim. Biophys. Acta 2007, 1770, 360–375. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.V. On the recognition of mammalian microsomal cytochrome P450 substrates and their characteristics—Towards the prediction of human P450 substrate specificity and metabolism. Biochem. Pharmacol. 2000, 60, 293–306. [Google Scholar] [CrossRef]
- Smith, D.A.; Ackland, M.J.; Jones, B.C. Properties of cytochrome P450 isoenzymes and their substrates.1. active site characteristics. Drug Discov. Today 1997, 2, 406–414. [Google Scholar] [CrossRef]
- Lewis, D.F.; Jacobs, M.N.; Dickins, M. Compound lipophilicity for substrate binding to human P450s in drug metabolism. Drug Discov. Today 2004, 9, 530–537. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Z.-R.; Ai, C.-Z.; Ge, G.-B.; He, Y.-Q.; Wu, J.-J.; Wang, J.-Y.; Man, H.-Z.; Jia, Y.; Yang, L. A Mechanism-Based Model for the Prediction of the Metabolic Sites of Steroids Mediated by Cytochrome P450 3A4. Int. J. Mol. Sci. 2015, 16, 14677-14694. https://doi.org/10.3390/ijms160714677
Dai Z-R, Ai C-Z, Ge G-B, He Y-Q, Wu J-J, Wang J-Y, Man H-Z, Jia Y, Yang L. A Mechanism-Based Model for the Prediction of the Metabolic Sites of Steroids Mediated by Cytochrome P450 3A4. International Journal of Molecular Sciences. 2015; 16(7):14677-14694. https://doi.org/10.3390/ijms160714677
Chicago/Turabian StyleDai, Zi-Ru, Chun-Zhi Ai, Guang-Bo Ge, Yu-Qi He, Jing-Jing Wu, Jia-Yue Wang, Hui-Zi Man, Yan Jia, and Ling Yang. 2015. "A Mechanism-Based Model for the Prediction of the Metabolic Sites of Steroids Mediated by Cytochrome P450 3A4" International Journal of Molecular Sciences 16, no. 7: 14677-14694. https://doi.org/10.3390/ijms160714677
APA StyleDai, Z.-R., Ai, C.-Z., Ge, G.-B., He, Y.-Q., Wu, J.-J., Wang, J.-Y., Man, H.-Z., Jia, Y., & Yang, L. (2015). A Mechanism-Based Model for the Prediction of the Metabolic Sites of Steroids Mediated by Cytochrome P450 3A4. International Journal of Molecular Sciences, 16(7), 14677-14694. https://doi.org/10.3390/ijms160714677