
Protein Folding and Mechanisms of Proteostasis


Abstract
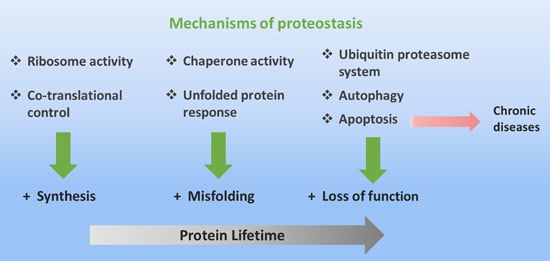
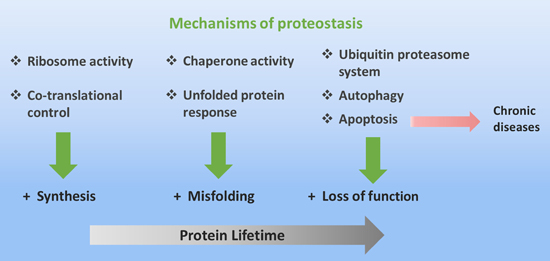
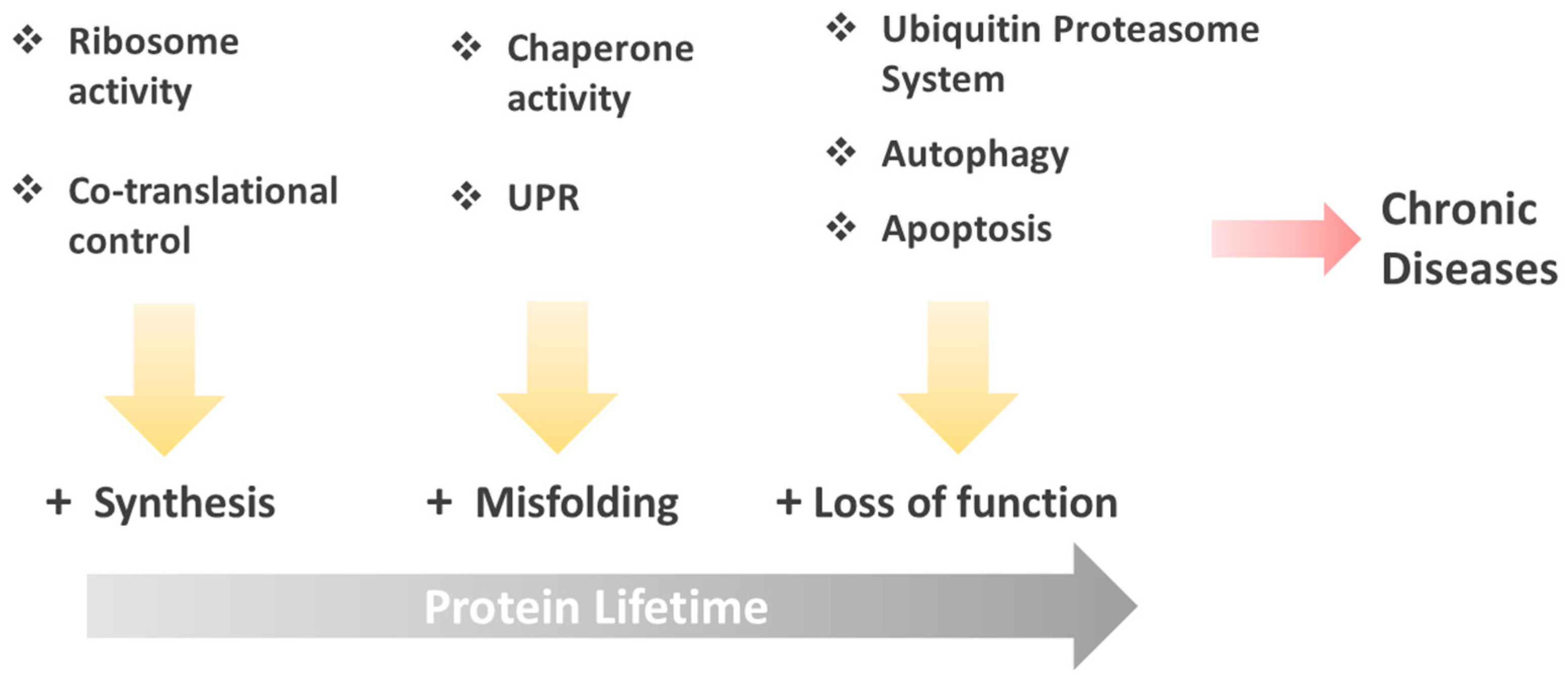
:
{kind=link}
{kind=link}
{kind=link}
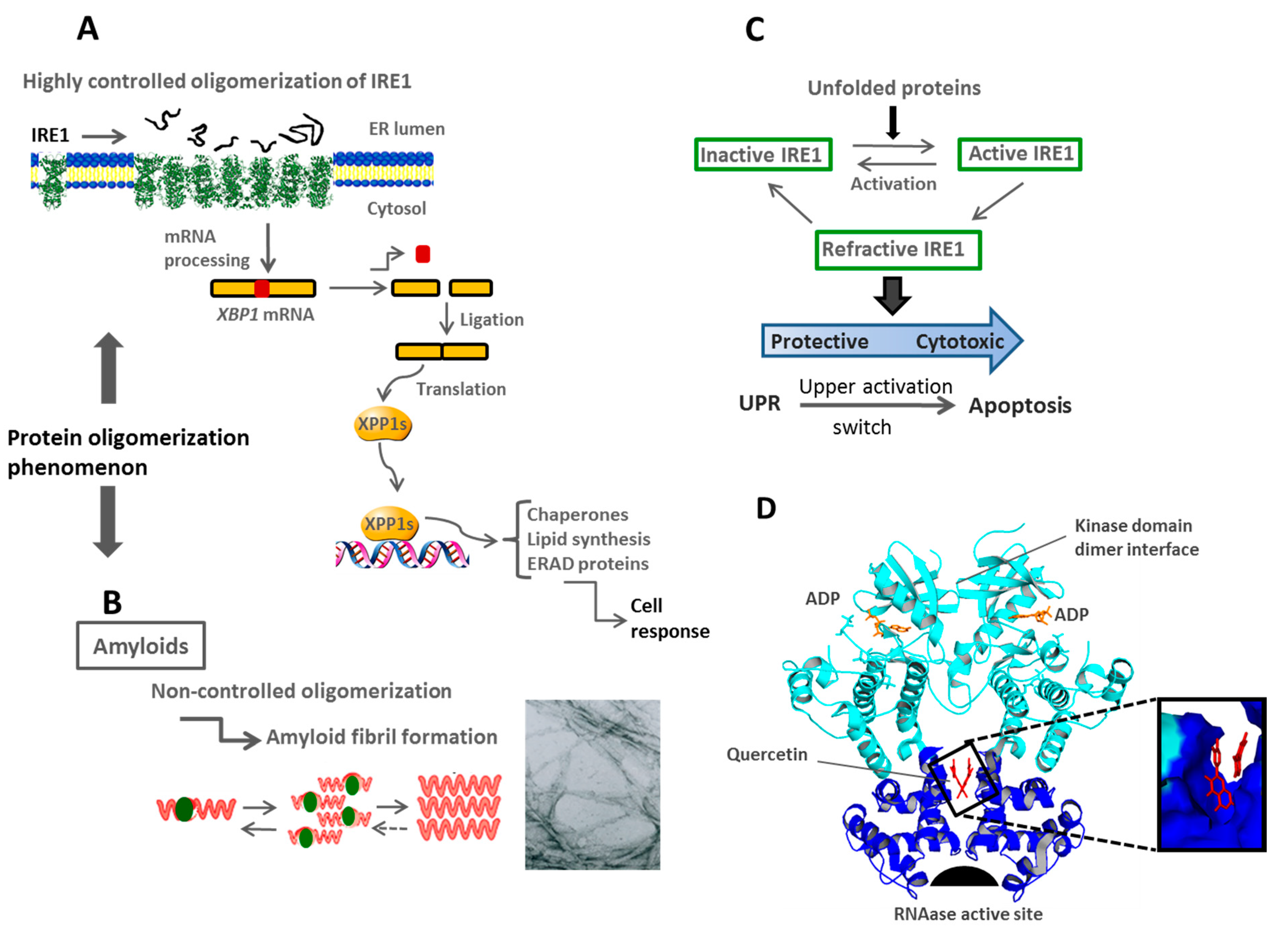{kind=link}
{kind=link}
1. Introduction
2. Protein Structure
2.1. Protein Folding
2.2. Allosteric Approach to Proteins
2.3. Partially Folded States in Proteins
2.4. Amyloid Focus

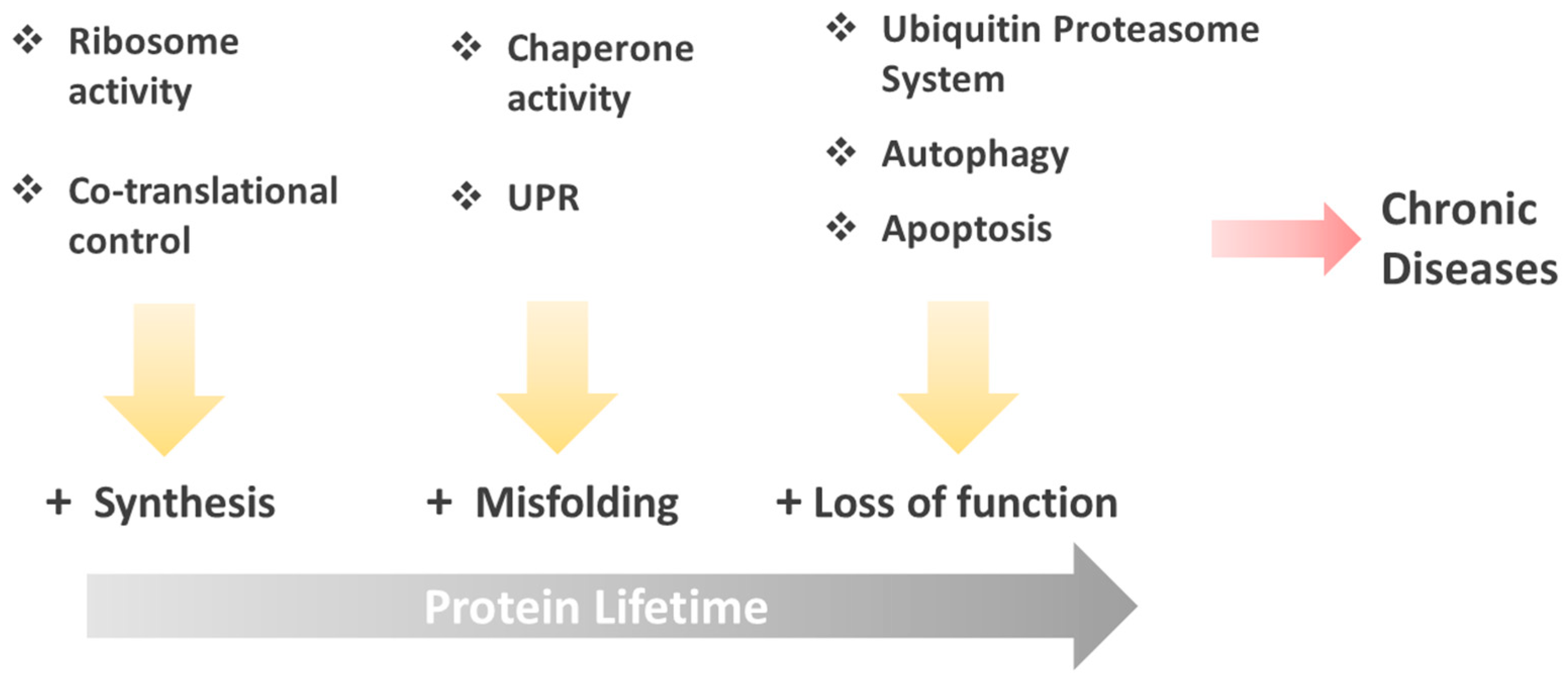
3. Mechanisms to Conserve Protein Structure
3.1. Chaperone Protein Function
3.2. Endoplasmic Reticulum Stress
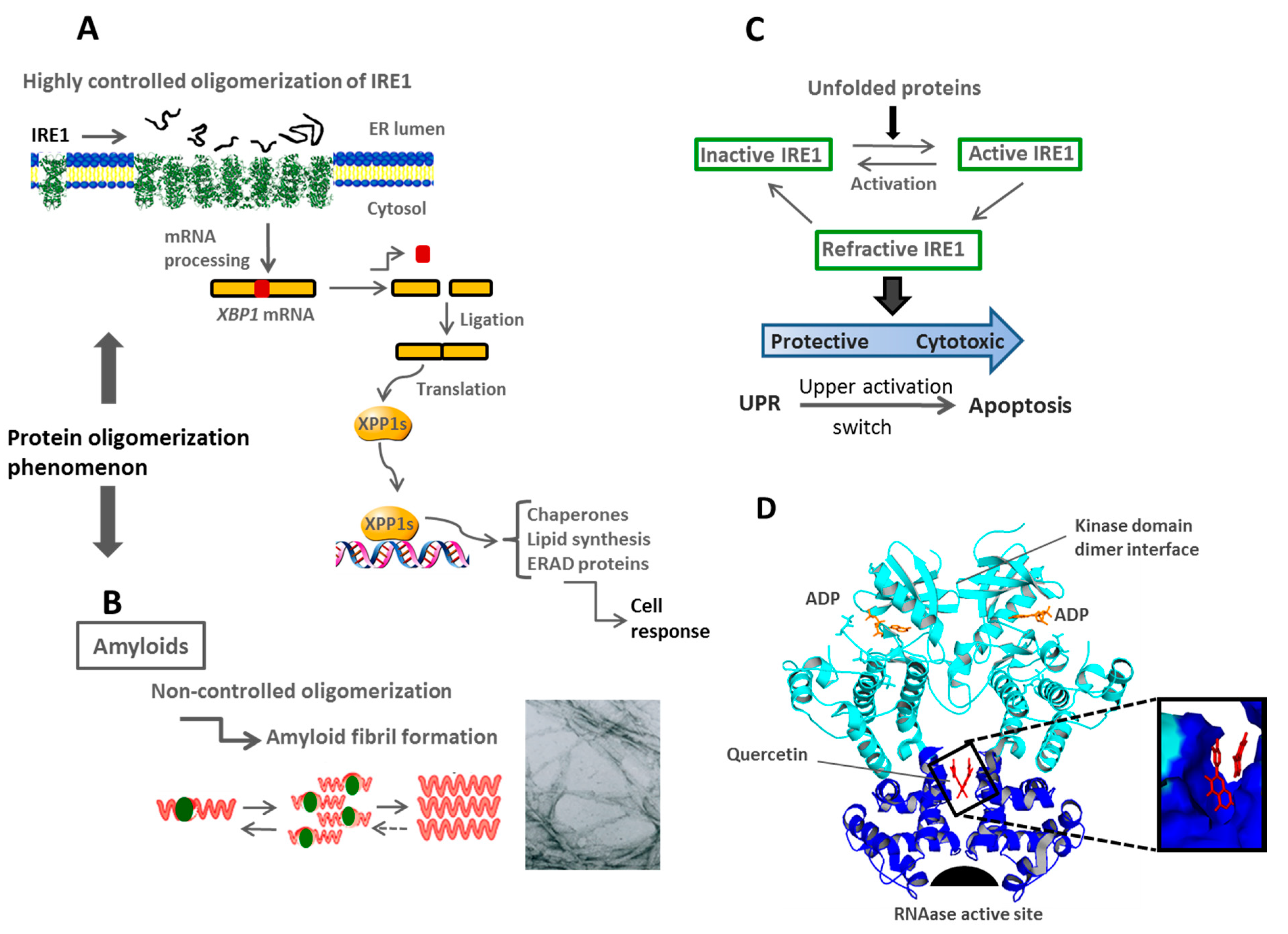
3.3. Spatial Compartmentalization of Protein Folding
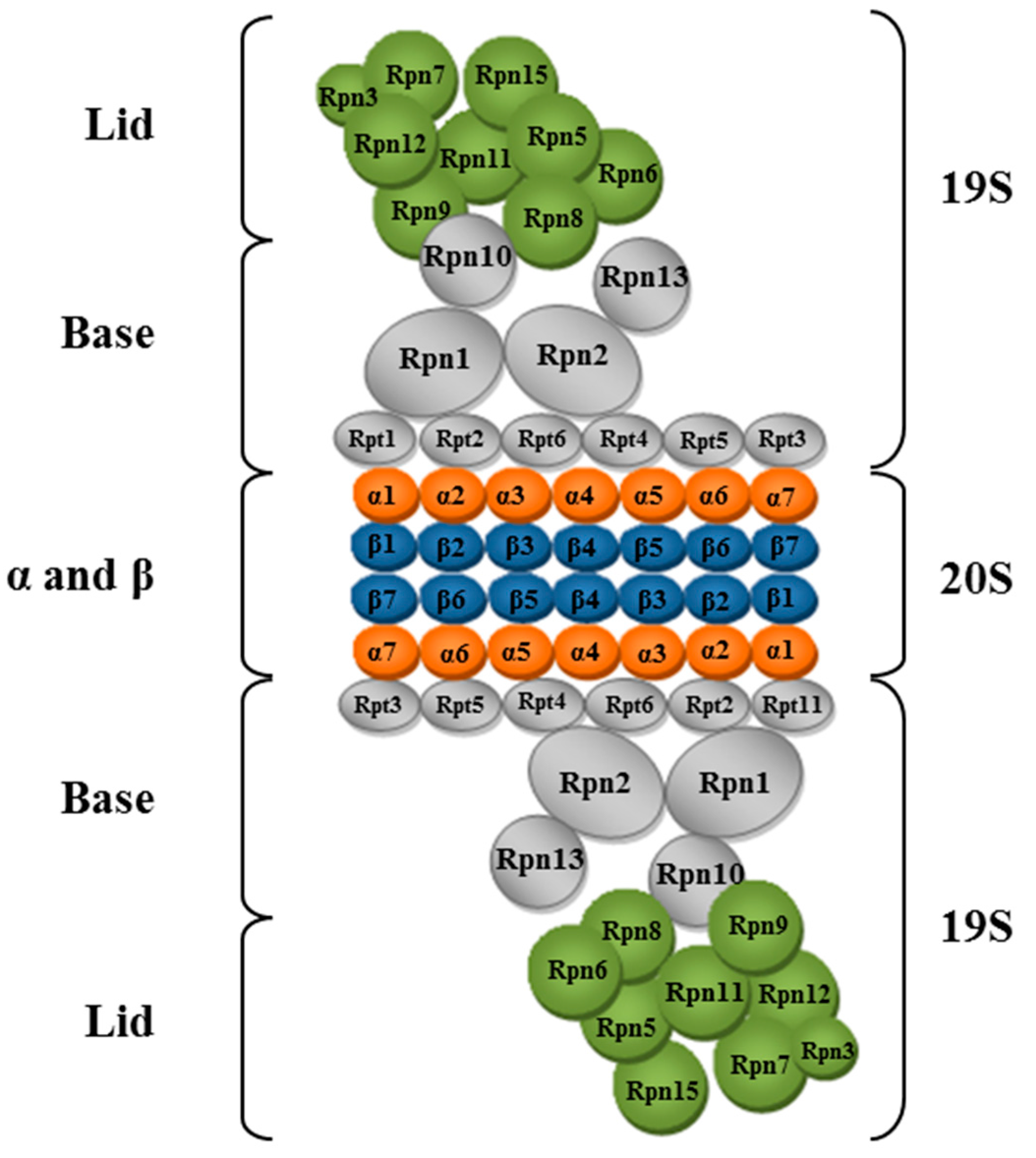
3.4. Proteasome, Structure, and Function

3.5. Autophagy Mechanism
4. Clinical Focus
4.1. Neurodegenerative Diseases
4.2. Metabolic Diseases
4.3. Cancer and Protein p53
5. Protein Folding in Drug Development
5.1. Strategies Focus on Amyloidosis
5.2. Chemical Chaperones
5.3. Final Considerations
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Badyaev, A.V. Stress-induced variation in evolution: From behavioural plasticity to genetic assimilation. Proc. Biol. Sci. 2005, 272, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R. The spatial structure of cell signaling systems. Phys. Biol. 2013, 10, 045004. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Wolynes, P.G. A second molecular biology revolution? The energy landscapes of biomolecular function. Phys. Chem. Chem. Phys. 2014, 16, 6321–6322. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Hegde, R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010, 189, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef]
- Seong, S.Y.; Matzinger, P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004, 4, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.; Chang, I.; Mohanty, S.; Luheshi, L.M.; Dobson, C.M.; Vendruscolo, M.; Favrin, G. Structural reorganisation and potential toxicity of oligomeric species formed during the assembly of amyloid fibrils. PLoS Comput. Biol. 2007, 3, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Ganzinger, K.A.; McColl, J.; Weimann, L.; Meehan, S.; Qamar, S.; Carver, J.A.; Wilson, M.R.; St George-Hyslop, P.; Dobson, C.M.; et al. Single molecule characterization of the interactions between amyloid-β peptides and the membranes of hippocampal cells. J. Am. Chem. Soc. 2013, 135, 1491–1498. [Google Scholar] [PubMed]
- Liu, Y.; Ye, Y. Proteostasis regulation at the endoplasmic reticulum: A new perturbation site for targeted cancer therapy. Cell Res. 2011, 21, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Princiotta, M.F.; Finzi, D.; Qian, S.B.; Gibbs, J.; Schuchmann, S.; Buttgereit, F.; Bennink, J.R.; Yewdell, J.W. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 2003, 18, 343–354. [Google Scholar] [CrossRef]
- Zhong, F.; Yang, D.; Hao, Y.; Lin, C.; Jiang, Y.; Ying, W.; Wu, S.; Zhu, Y.; Liu, S.; Yang, P.; et al. Regular patterns for proteome-wide distribution of protein abundance across species. PLoS ONE 2012, 7, e32423. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.E.; Marciniak, S.J. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 2. Protein misfolding and ER stress. Am. J. Physiol. Cell Physiol. 2014, 307, C657–C670. [Google Scholar] [CrossRef] [PubMed]
- Manié, S.N.; Lebeau, J.; Chevet, E. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 3. Orchestrating the unfolded protein response in oncogenesis: An update. Am. J. Physiol. Cell Physiol. 2014, 307, C901–C907. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Hinault, M.P.; Ben-Zvi, A.; Goloubinoff, P. Chaperones and proteases: Cellular fold-controlling factors of proteins in neurodegenerative diseases and aging. J. Mol. Neurosci. 2006, 30, 249–265. [Google Scholar] [CrossRef]
- Huang, K. Lectures on Statistical Physics and Protein Folding, 1st ed.; World Scientific: Hackensack, NJ, USA, 2005; pp. 1–100. [Google Scholar]
- Dobson, M.C. Protein chemistry: In the footsteps of alchemists. Science 2004, 304, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. Principles that govern folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Vuillon, L.; Lesieur, C. From local to global changes in proteins: A network view. Curr. Opin. Struct. Biol. 2015, 31, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J. Allostery in disease and in drug discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.; Hopkins, A. Navigating chemical space for biology and medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Zuiderweg, E.R.; Bertelsen, E.B.; Rousaki, A.; Mayer, M.P.; Gestwicki, J.E.; Ahmad, A. Allostery in the Hsp70 chaperone proteins. Top. Curr. Chem. 2013, 328, 99–153. [Google Scholar] [PubMed]
- Tompa, P. Multisteric regulation by structural disorder in modular signaling proteins: An extension of the concept of allostery. Chem. Rev. 2014, 114, 6715–6732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, S.; Pfohl, T. Real-time dynamics of emerging actin networks in cell-mimicking compartments. PLoS ONE 2015, 10, e011652. [Google Scholar] [CrossRef] [PubMed]
- Manz, B.N.; Groves, J.T. Spatial organization and signal transduction at intercellular junctions. Nat. Rev. Mol. Cell Biol. 2010, 11, 342–352. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006, 2, e88. [Google Scholar] [CrossRef] [PubMed]
- Verdian Doghaei, A.; Housaindokht, M.R.; Bozorgmehr, M.R. Molecular crowding effects on conformation and stability of G-quadruplex DNA structure: Insights from molecular dynamics simulation. J. Theor. Biol. 2015, 364, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, A.J. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Espinosa, P.; García-González, V.; Moreno, A.; Castillo, R.; Mas-Oliva, J. Disorder-to-order conformational transitions in protein structure and its relationship to disease. Mol. Cell. Biochem. 2009, 330, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, J.C.; Jakob, U. Conditional disorder in chaperone action. Trends Biochem. Sci. 2012, 37, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Górecki, A.; Bonarek, P.; Górka, A.K.; Figiel, M.; Wilamowski, M.; Dziedzicka-Wasylewska, M. Intrinsic disorder of human Yin Yang 1 protein. Proteins 2015. [Google Scholar] [CrossRef] [PubMed]
- García-González, V.; Mas-Oliva, J. Amyloidogenic properties of a D/N mutated 12 amino acid fragment of the C-terminal domain of the Cholesteryl-Ester Transfer Protein (CETP). Int. J. Mol. Sci. 2011, 12, 2019–2035. [Google Scholar] [CrossRef] [PubMed]
- Guharoy, M.; Pauwels, K.; Tompa, P. SnapShot: Intrinsic structural disorder. Cell 2015, 161, 1230–1230. [Google Scholar] [CrossRef] [PubMed]
- Forman-Kay, J.D.; Mittag, T. From sequence and forces to structure, function, and evolution of intrinsically disordered proteins. Structure 2013, 21, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- García-González, V.G.; Mas-Oliva, J. El Concepto de Enfermedad Asociado a la Conformación de Proteínas, 1st ed.; Universidad Nacional Autónoma de México and El Manual Moderno: Mexico City, Mexico, 2012; pp. 11–21. [Google Scholar]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M.; Oldfield, C.J.; Simon, I.; Dunker, A.K.; Uversky, V.N. Close encounters of the third kind: Disordered domains and the interactions of proteins. BioEssays 2009, 31, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The database of disordered proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed]
- Tantos, A.; Han, K.H.; Tompa, P. Intrinsic disorder in cell signaling and gene transcription. Mol. Cell. Endocrinol. 2012, 348, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Hilser, V.J.; Thompson, E.B. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 8311–8315. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.; Ferreon, J.C.; Wright, P.E.; Deniz, A.A. Modulation of allostery by protein intrinsic disorder. Nature 2013, 498, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Szász, C.; Buday, L. Structural disorder throws new light on moonlighting. Trends Biochem. Sci. 2005, 30, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Gould, C.M.; Diella, F.; Via, A.; Puntervoll, P.; Gemünd, C.; Chabanis-Davidson, S.; Michael, S.; Sayadi, A.; Bryne, J.C.; Chica, C.; et al. ELM: The status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 2010, 38, D167–D180. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Aloy, P. Contextual specificity in peptide-mediated protein interactions. PLoS ONE 2008, 3, e2524. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Lobley, A.; Swindells, M.B.; Orengo, C.A.; Jones, D.T. Inferring function using patterns of native disorder in proteins. PLoS Comput. Biol. 2007, 3, e162. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 473–484. [Google Scholar]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 2008, 9, S1. [Google Scholar] [CrossRef] [PubMed]
- Andresen, C.; Helander, S.; Lemak, A.; Farès, C.; Csizmok, V.; Carlsson, J.; Penn, L.Z.; Forman-Kay, J.D.; Arrowsmith, C.H.; Lundström, P.; et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 2012, 40, 6353–6266. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- James, C.; Tawfik, D.S. Conformational diversity and protein evolution—A 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003, 28, 361–368. [Google Scholar] [CrossRef]
- Bustos, D.M.; Iglesias, A.A. Intrinsic disorder is a key characteristic in partners that bind 14-3-3 proteins. Proteins 2006, 63, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 11504–11509. [Google Scholar] [CrossRef] [PubMed]
- Dalal, S.; Regan, L. Understanding the sequence determinants of conformational switching using protein design. Protein Sci. 2000, 9, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Mueller, J.P.; Van, Y.Y.; Romero, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006, 2, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Sudhof, T.C. A broken α-helix in folded α-synuclein. J. Biol. Chem. 2003, 278, 15313–15318. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold. Des. 1998, 3, R9–R23. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Meredith, S.C. Protein denaturation and aggregation: Cellular responses to denatured and aggregated proteins. Ann. N. Y. Acad. Sci. 2005, 1066, 181–221. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Fitzpatrick, A.W.; Meehan, S.; Mott, H.R.; Vendruscolo, M.; Dobson, C.M.; Welland, M.E. Role of intermolecular forces in defining material properties of protein nanofibrils. Science 2007, 318, 1900–1903. [Google Scholar] [CrossRef] [PubMed]
- De Genst, E.; Chirgadze, D.Y.; Klein, F.A.; Butler, D.C.; Matak-Vinković, D.; Trottier, Y.; Huston, J.S.; Messer, A.; Dobson, C.M. Structure of a single-chain fv bound to the 17 N-terminal residues of huntingtin provides insights into pathogenic amyloid formation and suppression. J. Mol. Biol. 2015, 427, 2166–2178. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Riek, R. On the possible amyloid origin of protein folds. J. Mol. Biol. 2012, 421, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Carny, O.; Gazit, E. A model for the role of short self-assembled peptides in the very early stages of the origin of life. FASEB J. 2005, 19, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid—From bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Fandrich, M.; Dobson, C.M. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 2002, 21, 5682–5690. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Claessen, D.; Rink, R.; de Jong, W.; Siebring, J.; de Vreugd, P.; Boersma, F.G.; Dijkhuizen, L.; Wosten, H.A. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003, 17, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Mackay, J.P.; Matthews, J.M.; Winefield, R.D.; Mackay, L.G.; Haverkamp, R.G.; Templeton, M.D. The hydrophobin EAS is largely unstructured in solution and functions by forming amyloid-like structures. Structure 2001, 9, 83–91. [Google Scholar] [CrossRef]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef] [PubMed]
- King, C.Y.; Tittmann, P.; Gross, H.; Gebert, R.; Aebi, M.; Wüthrich, K. Prion-inducing domain 2–114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 6618–6622. [Google Scholar] [CrossRef] [PubMed]
- Iconomidou, V.A.; Vriend, G.; Hamodrakas, S.J. Amyloids protect the silkmoth oocyte and embryo. FEBS Lett. 2000, 479, 141–145. [Google Scholar] [CrossRef]
- Iconomidou, V.A.; Chryssikos, G.D.; Gionis, V.; Galanis, A.S.; Cordopatis, P.; Hoenger, A.; Hamodrakas, S.J. Amyloid fibril formation propensity is inherent into the hexapeptide tandemly repeating sequence of the central domain of silkmoth chorion proteins of the A-family. J. Struct. Biol. 2006, 156, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Berson, J.F.; Theos, A.C.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol. 2003, 161, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Urabe, K.; Orlow, S.J.; Higashi, K.; Imokawa, G.; Kwon, B.S.; Potterf, B.; Hearing, V.J. The Pmel 17/silver locus protein. Characterization and investigation of its melanogenic function. J. Biol. Chem. 1994, 269, 29198–29205. [Google Scholar] [PubMed]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef] [PubMed]
- García-González, V.; Mas-Oliva, J. Amyloid fibril formation of peptides derived from the C-terminus of CETP modulated by lipids. Biochem. Biophys. Res. Commun. 2013, 434, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Escamilla, A.M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.P.; Dubay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Friendly and dangerous signals: Is the tissue in control? Nat. Immunol. 2007, 8, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M. Protein solubility and protein homeostasis: A generic view of protein misfolding disorders. Cold Spring Harb. Perspect. Biol. 2011, 3, a010454. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Calloni, G.; Chen, T.; Schermann, S.M.; Chang, H.-C.; Genevaux, P.; Agostini, F.; Tartaglia, G.G.; Hayer-Hartl, M.; Hartl, F.U. DnaK functions as a central hub in the E. coli chaperone network. Cell Rep. 2012, 1, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Willmund, F.; del Alamo, M.; Pechmann, S.; Chen, T.; Albanese, V.; Dammer, E.B.; Peng, J.; Frydman, J. The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell 2013, 152, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Gershenson, A.; Gierasch, L.M. Protein folding in the cell: Challenges and progress. Curr. Opin. Struct. Biol. 2011, 21, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Asherie, N. Protein crystallization and phase diagrams. Methods 2004, 34, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Finka, A.; Goloubinoff, P. Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones 2013, 18, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, D.; Xu, Y.; Cremers, C.M.; Ilbert, M.; Mittelman, R.; Fitzgerald, M.C.; Jakob, U. Order out of disorder: Working cycle of an intrinsically unfolded chaperone. Cell 2012, 148, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.C.; Fredrickson, E.K.; Oeser, M.L.; Garrett-Engele, C.M.; Locke, M.N.; Richardson, L.A.; Nelson, Z.W.; Hetrick, E.D.; Milac, T.I.; Gottschling, D.E.; et al. Disorder targets misorder in nuclear quality control degradation: A disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol. Cell 2011, 41, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P. Gymnastics of molecular chaperones. Mol. Cell 2010, 39, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Molecular chaperones: Assisting assembly in addition to folding. Trends Biochem. Sci. 2006, 31, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H.R. Chaperone machines in action. Curr. Opin. Struct. Biol. 2008, 18, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 chaperone machinery. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Tawfik, D.S. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature 2009, 459, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Slepenkov, S.V.; Witt, S.N. The unfolding story of the Escherichia coli Hsp70 DnaK: Is DnaK a holdase or an unfoldase? Mol. Microbiol. 2002, 45, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Bosl, B.; Grimminger, V.; Walter, S. The molecular chaperone Hsp104—A molecular machine for protein disaggregation. J. Struct. Biol. 2006, 156, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Langer, T.; Lu, C.; Echols, H.; Flanagan, J.; Hayer, M.K.; Hartl, F.U. Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature 1992, 356, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Horwich, A.L.; Fenton, W.A. Chaperonin-mediated protein folding: Using a central cavity to kinetically assist polypeptide chain folding. Q. Rev. Biophys. 2009, 42, 83–116. [Google Scholar] [CrossRef] [PubMed]
- Taldone, T.; Patel, H.J.; Bolaender, A.; Patel, M.R.; Chiosis, G. Protein chaperones: A composition of matter review (2008–2013). Expert Opin. Ther. Pat. 2014, 24, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Li, J.; Sha, B. Structural analysis of the Sil1-Bip complex reveals the mechanism for Sil1 to function as a nucleotide-exchange factor. Biochem. J. 2011, 438, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. The heat shock response: Systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb. Perspect. Biol. 2010, 2, a004390. [Google Scholar] [CrossRef] [PubMed]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M., Jr.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, D.; Tompa, P. Diverse functional manifestations of intrinsic structural disorder in molecular chaperones. Biochem. Soc. Trans. 2012, 40, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Snapp, E.L. Unfolded protein responses with or without unfolded proteins? Cells 2012, 1, 926–950. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Foit, L.; George, J.S.; Zhang, B.W.; Brooks, C.L., 3rd; Bardwell, J.C. Chaperone activation by unfolding. Proc. Natl. Acad. Sci. USA 2013, 110, E1254–E1262. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voeltz, G.K.; Rolls, M.M.; Rapoport, T.A. Structural organization of the endoplasmic reticulum. EMBO Rep. 2002, 3, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Shen, X.; Ellis, R.E.; Lee, K.; Liu, C.Y.; Yang, K.; Solomon, A.; Yoshida, H.; Morimoto, R.; Kurnit, D.M.; Mori, K.; et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 2001, 107, 893–903. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Stroud, R.M.; Zhang, C.; Shokat, K.M.; Walter, P. Cofactor-mediated conformational control in the bifunctional kinase/RNase Ire1. BMC Biol. 2011, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.W. Regulatory crosstalk within the mammalian unfolded protein response. Cell Mol. Life Sci. 2014, 71, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef] [PubMed]
- Escusa-Toret, S.; Vonk, W.I.; Frydman, J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol. 2013, 15, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Duttler, S.; Pechmann, S.; Frydman, J. Principles of cotranslational ubiquitination and quality control at the ribosome. Mol. Cell 2013, 50, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Durfee, L.A.; Huibregtse, J.M. A cotranslational ubiquitination pathway for quality control of misfolded proteins. Mol. Cell 2013, 50, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Pechmann, S.; Chartron, J.W.; Frydman, J. Local slowdown of translation by nonoptimal codons promotes nascent-chain recognition by SRP in vivo. Nat. Struct. Mol. Biol. 2014, 21, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Weissman, J.S.; Dillin, A. Differential scales of protein quality control. Cell 2014, 157, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.M.; Summers, D.W.; Cyr, D.M. Molecular chaperones antagonize proteotoxicity by differentially modulating protein aggregation pathways. Prion 2009, 3, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Salmonowicz, H.; Brown, R.; Turkowska, J.; Średniawa, W.; Pattabiraman, S.; Amen, T.; Abraham, A.C.; Eichler, N.; Lyakhovetsky, R.; et al. Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin. Proc. Natl. Acad. Sci. USA 2014, 11, 8049–8054. [Google Scholar] [CrossRef] [PubMed]
- Polling, S.; Mok, Y.F.; Ramdzan, Y.M.; Turner, B.J.; Yerbury, J.J.; Hill, A.F.; Hatters, D.M. Misfolded polyglutamine, polyalanine, and superoxide dismutase 1 aggregate via distinct pathways in the cell. J. Biol. Chem. 2014, 289, 6669–6690. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.J.; Lyakhovetsky, R.; Werdiger, A.-C.; Gitler, A.D.; Soen, Y.; Kaganovich, D. Compartmentalization of superoxide dismutase 1 (SOD1G93A) aggregates determines their toxicity. Proc. Natl. Acad. Sci. USA 2012, 109, 15811–15816. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.; Arosio, P.; Presto, J.; Kurudenkandy, F.R.; Biverstål, H.; Dolfe, L.; Dunning, C.; Yang, X.; Frohm, B.; Vendruscolo, M.; et al. A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 2015, 22, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.H. Protein folding: Misfolded proteins join the Q. Nat. Rev. Mol. Cell Biol. 2013, 14, 608. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Voisine, C.; Pedersen, J.S.; Morimoto, R.I. Chaperone networks: Tipping the balance in protein folding diseases. Neurobiol. Dis. 2010, 40, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, E.K.; Gardner, R.G. Selective destruction of abnormal proteins by ubiquitin-mediated protein quality control degradation. Semin. Cell Dev. Biol. 2012, 23, 530–537. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Kusmierczyk, A.R.; Kunjappu, M.J.; Funakoshi, M.; Hochstrasser, M. A multimeric assembly factor controls the formation of alternative 20S proteasomes. Nat. Struct. Mol. Biol. 2008, 15, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Konstantinova, I.M.; Tsimokha, A.S.; Mittenberg, A.G.; Kwang, W.J. Role of proteasomes in cellular regulation. In International Review of Cell and Molecular Biology; Academic Press: San Diego, CA, USA, 2008; Volume 267, pp. 59–124. [Google Scholar]
- Da Fonseca, P.C.A.; He, J.; Morris, E.P. Molecular model of the human 26S proteasome. Mol. Cell 2012, 46, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Osmulski, P.A.; Gaczynska, M.; Glickman, M.H. The central unit within the 19S regulatory particle of the proteasome. Nat. Struct. Mol. Biol. 2008, 15, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Iwafune, Y.; Akiyama, T.; Okayama, A.; Nakamura, H.; Arakawa, N.; Kimura, Y.; Hirano, H. Co- and post-translational modifications of the 26S proteasome in yeast. Proteomics 2010, 10, 2769–2779. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B 2009, 85, 12–36. [Google Scholar] [CrossRef]
- Li, X.; Demartino, G.N. Variably modulated gating of the 26S proteasome by ATP and polyubiquitin. Biochem. J. 2009, 421, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Sledź, P.; Förster, F.; Baumeister, W.J. Allosteric effects in the regulation of 26S proteasome activities. Mol. Biol. 2013, 425, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Davis, P.B.; Morris, J.C.; White, D.L. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol. Aging 1991, 12, 295–312. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 198, 82, 4245–4259. [Google Scholar]
- Gregori, L.; Hainfeld, J.F.; Simon, M.N.; Goldgaber, D. Binding of amyloid β protein to the 20 S proteasome. J. Biol. Chem. 1997, 272, 58–62. [Google Scholar] [PubMed]
- Gregori, L.; Fuchs, C.; Figueiredo-Pereira, M.E.; van Nostrand, W.E.; Goldgaber, D. Amyloid β-protein inhibits ubiquitin-dependent protein degradation in vitro. J. Biol. Chem. 1995, 270, 19702–19708. [Google Scholar] [CrossRef] [PubMed]
- Lopez Salon, M.; Pasquini, L.; Besio Moreno, M.; Pasquini, J.M.; Soto, E. Relationship between β-amyloid degradation and the 26S proteasome in neural cells. Exp. Neurol. 2003, 180, 131–143. [Google Scholar] [CrossRef]
- Oh, S.; Hong, H.S.; Hwang, E.; Sim, H.J.; Lee, W.; Shin, S.J.; Mook-Jung, I. Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech. Ageing Dev. 2005, 126, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Hill, J.A. Protein quality control and metabolism: Bidirectional control in the heart. Cell Metab. 2015, 21, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Klionsky, D.J. Autophagic processes in yeast: Mechanism, machinery and regulation. Genetics 2013, 194, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Inagaki, F. Mechanisms of autophagy. Annu. Rev. Biophys. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy a novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, E.; Cecconi, F. Autophagic and apoptotic response to stress signals in mammalian cells. Arch. Biochem. Biophys. 2007, 462, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Gavrin, L.K.; Denny, R.A.; Saiah, E. Small molecules that target protein misfolding. J. Med. Chem. 2012, 55, 10823–10843. [Google Scholar] [CrossRef] [PubMed]
- Ciryam, P.; Tartaglia, G.G.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep. 2013, 5, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Brocchieri, L.; Karlin, S. Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res. 2005, 33, 3390–3400. [Google Scholar] [CrossRef] [PubMed]
- Ogle, J.M.; Ramakrishnan, V. Structural insights into translational fidelity. Annu. Rev. Biochem. 2005, 74, 129–177. [Google Scholar] [CrossRef] [PubMed]
- Pechmann, S.; Willmund, F.; Frydman, J. The ribosome as a hub for protein quality control. Mol. Cell 2013, 49, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; Shu, W.; Devlin, G.L.; Meehan, S.; Auer, S.; Dobson, C.M.; Welland, M.E. Kinetics and thermodynamics of amyloid formation from direct measurements of fluctuations in fibril mass. Proc. Natl. Acad. Sci. USA 2007, 104, 10016–10021. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.Q.; Wu, S.; Buell, A.K.; Cohen, S.I.; Chen, L.J.; Hu, W.H.; Cusack, S.A.; Itzhaki, L.S.; Zhang, H.; Knowles, T.P.; et al. Influence of specific Hsp70 domains on fibril formation of the yeast prion protein Ure2. Philos. Trans. R. Soc. B 2013, 368, 20110410. [Google Scholar]
- Lee, J.; Ozcan, U. Unfolded protein response signaling and metabolic diseases. J. Biol. Chem. 2014, 289, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Stultz, C.M. Finding order within disorder: Elucidating the structure of proteins associated with neurodegenerative disease. Future Med. Chem. 2009, 1, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Igoillo-Esteve, M.; Cunha, D.A.; Ladrière, L.; Eizirik, D.L. An update on lipotoxic endoplasmic reticulum stress in pancreatic β-cells. Biochem. Soc. Trans. 2008, 36, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Haass, C. Intramembrane proteolysis by presenilins. Nat. Rev. Mol. Cell Biol. 2000, 1, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Arnold, B.; Cassady, S.J.; Chu, C.T.; Burton, E.A.; Berman, S.B. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum. Mol. Genet. 2011, 20, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Targeting endoplasmic reticulum stress in metabolic disease. Expert Opin. Ther. Targets 2012, 17, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Abedini, A.; Raleigh, D.P. Aggregation of islet amyloid polypeptide: From physical chemistry to cell biology. Curr. Opin. Struct. Biol. 2013, 23, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.S. Human IAPP amyloidogenic properties and pancreatic β-cell death. Cell Calcium 2014, 56, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Casas, S.; Gomis, R.; Gribble, F.M.; Altirriba, J.; Knuutila, S.; Novials, A. Impairment of the ubiquitin-proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic β-cell apoptosis. Diabetes 2007, 56, 2284–2294. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Huang, C.J.; Gurlo, T.; Daval, M.; Matveyenko, A.V.; Rizza, R.A.; Butler, A.E.; Butler, P.C. β-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes 2011, 60, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Welters, H.J.; Tadayyon, M.; Scarpello, J.H.; Smith, S.A.; Morgan, N.G. Mono-unsaturated fatty acids protect against β-cell apoptosis induced by saturated fatty acids, serum withdrawal or cytokine exposure. FEBS Lett. 2004, 560, 103–108. [Google Scholar] [CrossRef]
- Díaz-Villanueva, J.F.; García-González, V. Key interactions in the binding of free fatty acids on amylin-structure transitions. 2015; manuscript in preparation. [Google Scholar]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lerner, A.G.; Vande, W.L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Chin, K.T.; Zito, E.; Zhang, Y.; Heldman, N.; Harding, H.P.; Fass, D.; Thorpe, C.; Ron, D.A. Small molecule inhibitor of endoplasmic reticulum oxidation 1 (ERO1) with selectively reversible thiol reactivity. J. Biol. Chem. 2010, 285, 20993–21003. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10631–10634. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Banh, A.; Papandreou, I.; Cao, H.; Galvez, M.G.; Gurtner, G.C.; Denko, N.C.; Le, Q.T.; Koong, A.C. Imaging the unfolded protein response in primary tumors reveals microenvironments with metabolic variations that predict tumor growth. Cancer Res. 2010, 70, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef] [PubMed]
- Pyrko, P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C. GRP78/BiP modulation of GRP78/BiP in altering sensitivity to chemotherapy. Methods Enzymol. 2011, 491, 25–36. [Google Scholar] [PubMed]
- Li, X.; Zhang, K.; Li, Z. Unfolded protein response in cancer: The physician’s perspective. J. Hematol. Oncol. 2011, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Park, H.R.; Tomida, A.; Sato, S.; Tsukumo, Y.; Yun, J.; Yamori, T.; Hayakawa, Y.; Tsuruo, T.; Shin-ya, K. Effect on tumor cells of blocking survival response to glucose deprivation. J. Natl. Cancer Inst. 2004, 96, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Furuno, A.; Sakurai, J.; Sakamoto, A.; Park, H.R.; Shin-Ya, K.; Tsuruo, T.; Tomida, A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res. 2009, 69, 4225–4234. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M.; Krivoshein, A.V.; Hamby, C.V.; Pizzonia, J.; Gilbert, K.S.; Ray, Y.S.; Brand, H.; Paton, A.W.; Paton, J.C.; Backer, M.V. Chaperone-targeting cytotoxin and endoplasmic reticulum stress-inducing drug synergize to kill cancer cells. Neoplasia 2009, 11, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 12, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 1, a001008. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Segalat, L. Loss-of-function genetic diseases and the concept of pharmaceutical targets. Orphanet. J. Rare Dis. 2007, 2, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Rippin, T.M.; Bykov, V.J.; Freund, S.M.; Selivanova, G.; Wiman, K.G.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Friedler, A.; Hansson, L.O.; Veprintsev, D.B.; Freund, S.M.; Rippin, T.M.; Nikolova, P.V.; Proctor, M.R.; Rüdiger, S.; Fersht, A.R. A peptide that binds and stabilizes p53 core domain: Chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. USA 2002, 99, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Macromolecular crowding: An important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 2001, 11, 114–119. [Google Scholar] [CrossRef]
- Gribbin, J.R. Deep Simplicity: Bringing Order to Chaos and Complexity, 1st ed.; Random House Inc.: New York, NY., USA, 2004; pp. 1–304. [Google Scholar]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [PubMed]
- Perutz, M.F.; Pope, B.J.; Owen, D.; Wanker, E.E.; Scherzinger, E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine rich domains of Sup35 and of the amyloid β-peptide of amyloid plaques. Proc. Natl. Acad. Sci. USA 2002, 99, 5596–5600. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, J.I.; Sunde, M.; Jones, J.A.; Campbell, I.D.; Dobson, C.M. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. USA 1998, 95, 4224–4228. [Google Scholar] [CrossRef] [PubMed]
- Wasmer, C.; Schütz, A.; Loquet, A.; Buhtz, C.; Greenwald, J.; Riek, R.; Böckmann, A.; Meier, B.H. The molecular organization of the fungal prion HET-s in its amyloid form. J. Mol. Biol. 2009, 394, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Wiseman, R.L.; Sekijima, Y.; Green, N.S.; Adamski-Werner, S.L.; Kelly, J.W. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: A focus on the transthyretin amyloidoses. Acc. Chem. Res. 2005, 38, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Lansbury, P. Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease? Proc. Natl. Acad. Sci. USA 1999, 96, 3342–3344. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Kraskiewicz, H.; FitzGerald, U. InterfERing with endoplasmic reticulum stress. Trends Pharmacol. Sci. 2012, 33, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.L.; Kelly, J.W. Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: Progress and prognosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004507. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Giacca, A.; Lewis, G.F. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and β-cell dysfunction in humans. Diabetes 2011, 60, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Promlek, T.; Ishiwata-Kimata, Y.; Shido, M.; Sakuramoto, M.; Kohno, K.; Kimata, Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell 2011, 22, 3520–3532. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, R.L.; Zhang, Y.; Lee, K.P.; Harding, H.P.; Haynes, C.M.; Price, J.; Sicheri, F.; Ron, D. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol. Cell 2010, 38, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Villanueva, J.F.; Díaz-Molina, R.; García-González, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193-17230. https://doi.org/10.3390/ijms160817193
Díaz-Villanueva JF, Díaz-Molina R, García-González V. Protein Folding and Mechanisms of Proteostasis. International Journal of Molecular Sciences. 2015; 16(8):17193-17230. https://doi.org/10.3390/ijms160817193
Chicago/Turabian StyleDíaz-Villanueva, José Fernando, Raúl Díaz-Molina, and Victor García-González. 2015. "Protein Folding and Mechanisms of Proteostasis" International Journal of Molecular Sciences 16, no. 8: 17193-17230. https://doi.org/10.3390/ijms160817193