
Fumaric Acid Esters Do Not Reduce Inflammatory NF-κB/p65 Nuclear Translocation, ICAM-1 Expression and T-Cell Adhesiveness of Human Brain Microvascular Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
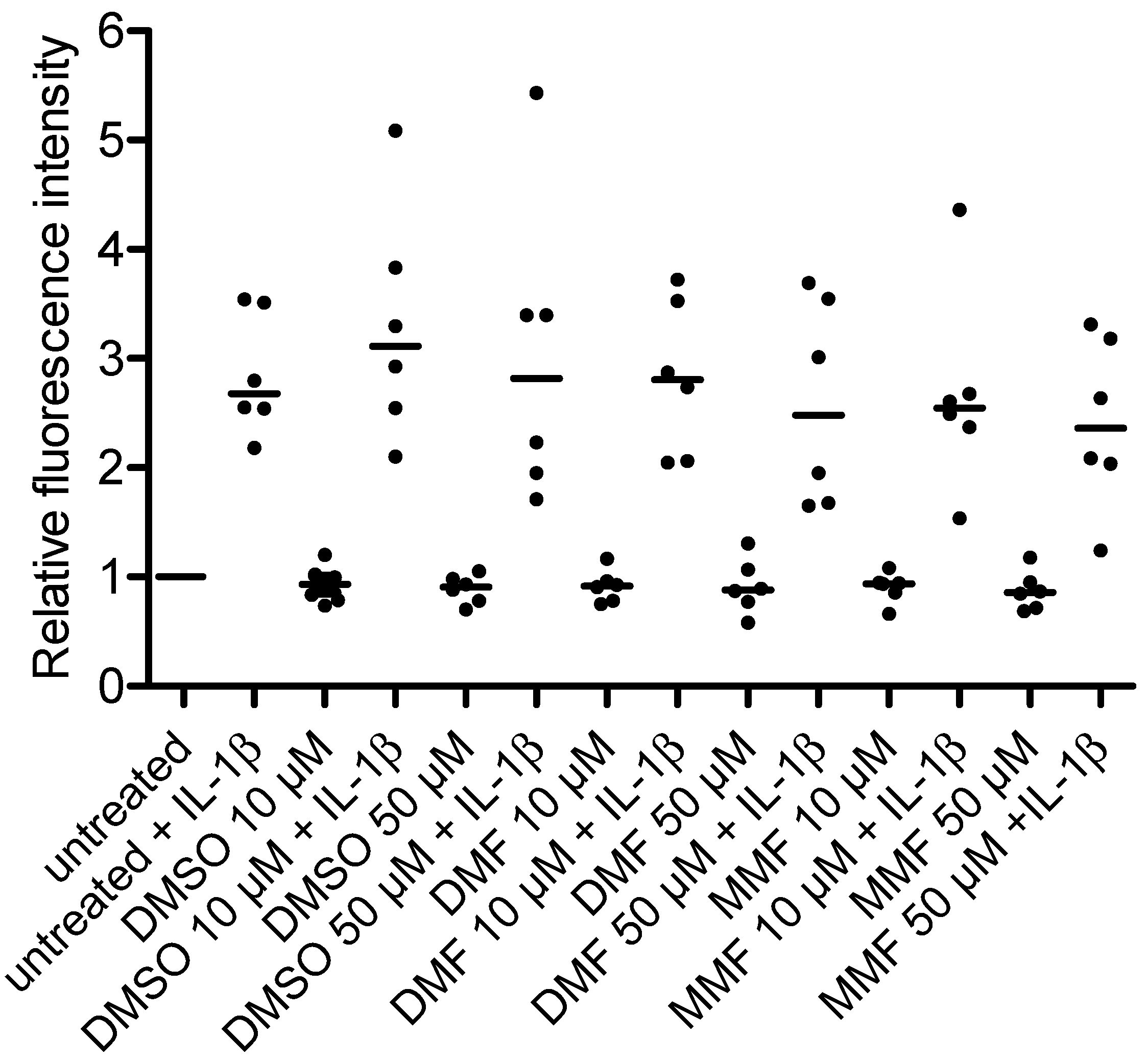
2.1. FAE Do Not Modulate NF-κB/p65 Nuclear Translocation or p38 MAPK Activation in HBMEC

2.2. FAE Do Not Reduce Basal or Inflammatory-Inducible Expression of ICAM-1 in HBMEC

2.3. FAE Do Not Reduce T-Cell Adhesiveness of HBMEC

3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Stimulation
4.3. Cell Protein Extracts and Western Blotting
4.4. Flow Cytometry
4.5. Adhesion Assay
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Engelhardt, B.; Ransohoff, R.M. Capture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends Immunol. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Buttmann, M.; Rieckmann, P. Treating multiple sclerosis with monoclonal antibodies. Expert Rev. Neurother. 2008, 8, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Haarmann, A.; Nowak, E.; Deiss, A.; van der Pol, S.; Monoranu, C.M.; Kooij, G.; Muller, N.; van der Valk, P.; Stoll, G.; de Vries, H.E.; et al. Soluble VCAM-1 impairs human brain endothelial barrier integrity via integrin α-4-transduced outside-in signalling. Acta. Neuropathol. 2015, 129, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Van Doorn, R.; Lopes Pinheiro, M.A.; Kooij, G.; Lakeman, K.; van het Hof, B.; van der Pol, S.M.; Geerts, D.; van Horssen, J.; van der Valk, P.; van der Kam, E.; et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J. Neuroinflamm. 2012, 9, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, S.; Goelz, S.; Linker, R.; Luehder, F.; Gold, R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol. 2006, 145, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Treumer, F.; Zhu, K.; Glaser, R.; Mrowietz, U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J. Investig. Dermatol. 2003, 121, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Dehmel, T.; Dobert, M.; Pankratz, S.; Leussink, V.I.; Hartung, H.P.; Wiendl, H.; Kieseier, B.C. Monomethylfumarate reduces in vitro migration of mononuclear cells. Neurol. Sci. 2014, 35, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Rubant, S.A.; Ludwig, R.J.; Diehl, S.; Hardt, K.; Kaufmann, R.; Pfeilschifter, J.M.; Boehncke, W.H. Dimethylfumarate reduces leukocyte rolling in vivo through modulation of adhesion molecule expression. J. Investig. Dermatol. 2008, 128, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Wallbrecht, K.; Drick, N.; Hund, A.C.; Schon, M.P. Downregulation of endothelial adhesion molecules by dimethylfumarate, but not monomethylfumarate, and impairment of dynamic lymphocyte-endothelial cell interactions. Exp. Dermatol. 2011, 20, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Vandermeeren, M.; Janssens, S.; Borgers, M.; Geysen, J. Dimethylfumarate is an inhibitor of cytokine-induced E-selectin, VCAM-1, and ICAM-1 expression in human endothelial cells. Biochem. Biophys. Res. Commun. 1997, 234, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Loewe, R.; Holnthoner, W.; Groger, M.; Pillinger, M.; Gruber, F.; Mechtcheriakova, D.; Hofer, E.; Wolff, K.; Petzelbauer, P. Dimethylfumarate inhibits TNF-induced nuclear entry of NF-κB/p65 in human endothelial cells. J. Immunol. 2002, 168, 4781–4787. [Google Scholar] [CrossRef] [PubMed]
- Kallmann, B.A.; Wagner, S.; Hummel, V.; Buttmann, M.; Bayas, A.; Tonn, J.C.; Rieckmann, P. Characteristic gene expression profile of primary human cerebral endothelial cells. FASEB J. 2002, 16, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Voraberger, G.; Schafer, R.; Stratowa, C. Cloning of the human gene for intercellular adhesion molecule 1 and analysis of its 5′-regulatory region. Induction by cytokines and phorbol ester. J. Immunol. 1991, 147, 2777–2786. [Google Scholar] [PubMed]
- Abadier, M.; Haghayegh Jahromi, N.; Cardoso Alves, L.; Boscacci, R.; Vestweber, D.; Barnum, S.; Deutsch, U.; Engelhardt, B.; Lyck, R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur. J. Immunol. 2015, 45, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Venci, J.V.; Gandhi, M.A. Dimethyl fumarate (Tecfidera): A new oral agent for multiple sclerosis. Ann. Pharmacother. 2013, 47, 1697–1702. [Google Scholar] [CrossRef] [PubMed]
- Benardais, K.; Pul, R.; Singh, V.; Skripuletz, T.; Lee, D.H.; Linker, R.A.; Gudi, V.; Stangel, M. Effects of fumaric acid esters on blood-brain barrier tight junction proteins. Neurosci. Lett. 2013, 555, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Kunze, R.; Urrutia, A.; Hoffmann, A.; Liu, H.; Helluy, X.; Pham, M.; Reischl, S.; Korff, T.; Marti, H.H. Dimethyl fumarate attenuates cerebral edema formation by protecting the blood-brain barrier integrity. Exp. Neurol. 2015, 266, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Gumbleton, M.; Audus, K.L. Progress and limitations in the use of in vitro cell cultures to serve as a permeability screen for the blood-brain barrier. J. Pharm. Sci. 2001, 90, 1681–1698. [Google Scholar] [CrossRef] [PubMed]
- Omidi, Y.; Campbell, L.; Barar, J.; Connell, D.; Akhtar, S.; Gumbleton, M. Evaluation of the immortalised mouse brain capillary endothelial cell line, b.End3, as an in vitro blood-brain barrier model for drug uptake and transport studies. Brain Res. 2003, 990, 95–112. [Google Scholar] [CrossRef]
- Song, L.; Pachter, J.S. Culture of murine brain microvascular endothelial cells that maintain expression and cytoskeletal association of tight junction-associated proteins. In Vitro Cell. Dev. Biol. Anim. 2003, 39, 313–320. [Google Scholar] [CrossRef]
- Haarmann, A.; Deiss, A.; Prochaska, J.; Foerch, C.; Weksler, B.; Romero, I.; Couraud, P.O.; Stoll, G.; Rieckmann, P.; Buttmann, M. Evaluation of soluble junctional adhesion molecule—A as a biomarker of human brain endothelial barrier breakdown. PLoS ONE 2010, 5, e13568. [Google Scholar] [CrossRef] [PubMed]
- Buttmann, M.; Lorenz, A.; Weishaupt, A.; Rieckmann, P. Atorvastatin partially prevents an inflammatory barrier breakdown of cultured human brain endothelial cells at a pharmacologically relevant concentration. J. Neurochem. 2007, 102, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Buttmann, M.; Berberich-Siebelt, F.; Serfling, E.; Rieckmann, P. Interferon-beta is a potent inducer of interferon regulatory factor-1/2-dependent IP-10/CXCL10 expression in primary human endothelial cells. J. Vasc. Res. 2007, 44, 51–60. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haarmann, A.; Nehen, M.; Deiß, A.; Buttmann, M. Fumaric Acid Esters Do Not Reduce Inflammatory NF-κB/p65 Nuclear Translocation, ICAM-1 Expression and T-Cell Adhesiveness of Human Brain Microvascular Endothelial Cells. Int. J. Mol. Sci. 2015, 16, 19086-19095. https://doi.org/10.3390/ijms160819086
Haarmann A, Nehen M, Deiß A, Buttmann M. Fumaric Acid Esters Do Not Reduce Inflammatory NF-κB/p65 Nuclear Translocation, ICAM-1 Expression and T-Cell Adhesiveness of Human Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences. 2015; 16(8):19086-19095. https://doi.org/10.3390/ijms160819086
Chicago/Turabian StyleHaarmann, Axel, Mathias Nehen, Annika Deiß, and Mathias Buttmann. 2015. "Fumaric Acid Esters Do Not Reduce Inflammatory NF-κB/p65 Nuclear Translocation, ICAM-1 Expression and T-Cell Adhesiveness of Human Brain Microvascular Endothelial Cells" International Journal of Molecular Sciences 16, no. 8: 19086-19095. https://doi.org/10.3390/ijms160819086
APA StyleHaarmann, A., Nehen, M., Deiß, A., & Buttmann, M. (2015). Fumaric Acid Esters Do Not Reduce Inflammatory NF-κB/p65 Nuclear Translocation, ICAM-1 Expression and T-Cell Adhesiveness of Human Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences, 16(8), 19086-19095. https://doi.org/10.3390/ijms160819086