
Current Proceedings in the Molecular Dissection of Hepatocellular Adenomas: Review and Hands-on Guide for Diagnosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Clinical Background


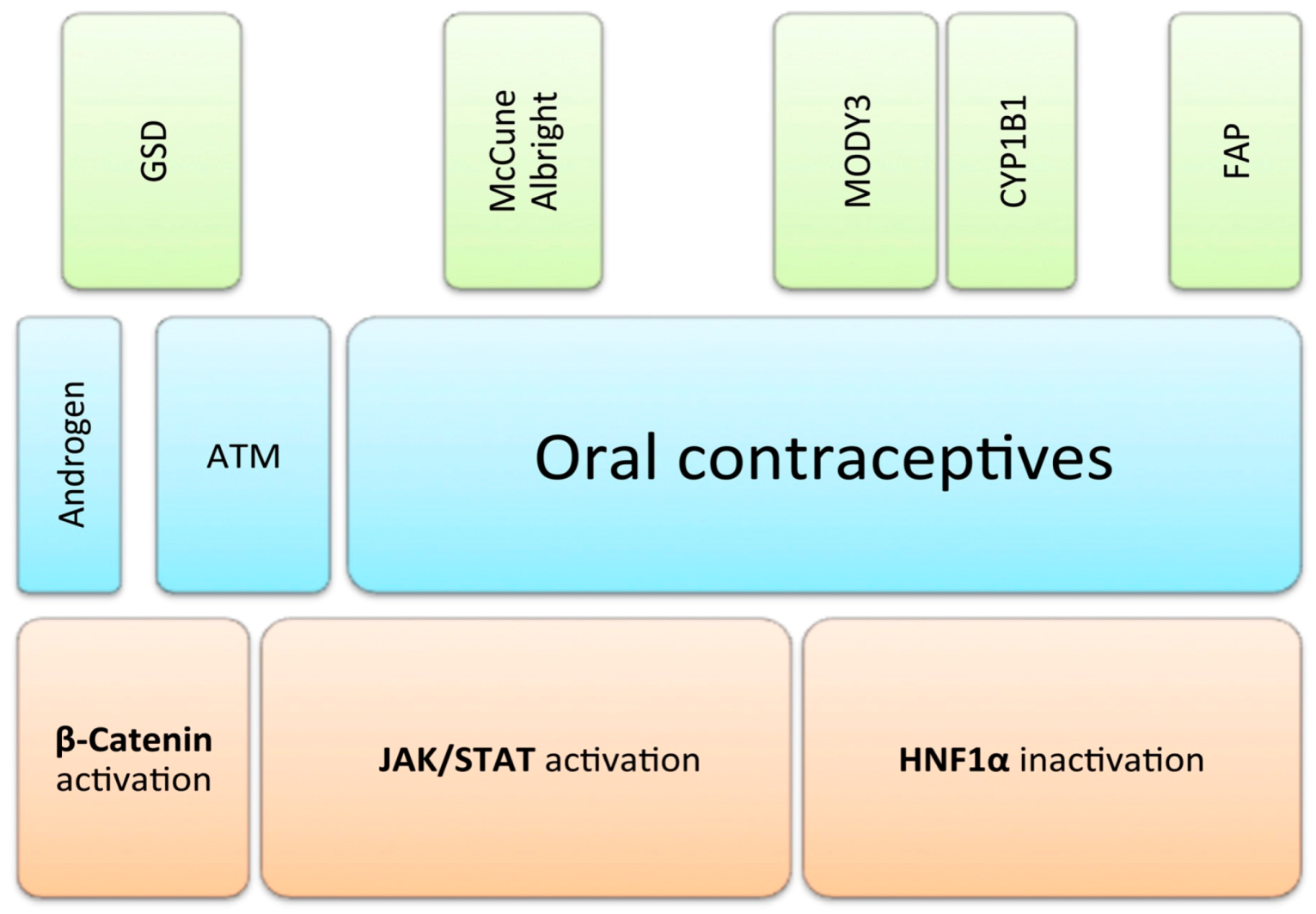
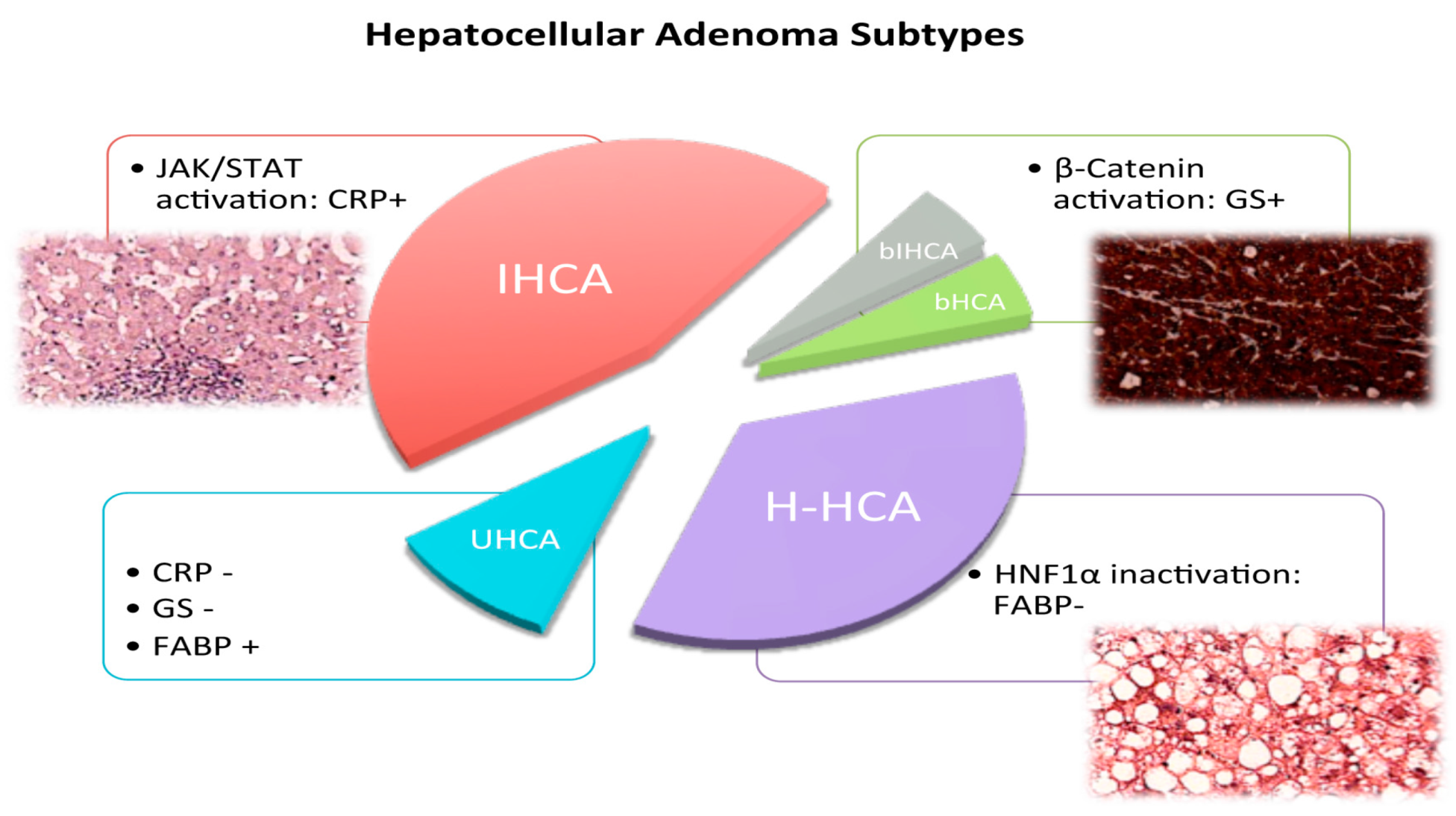
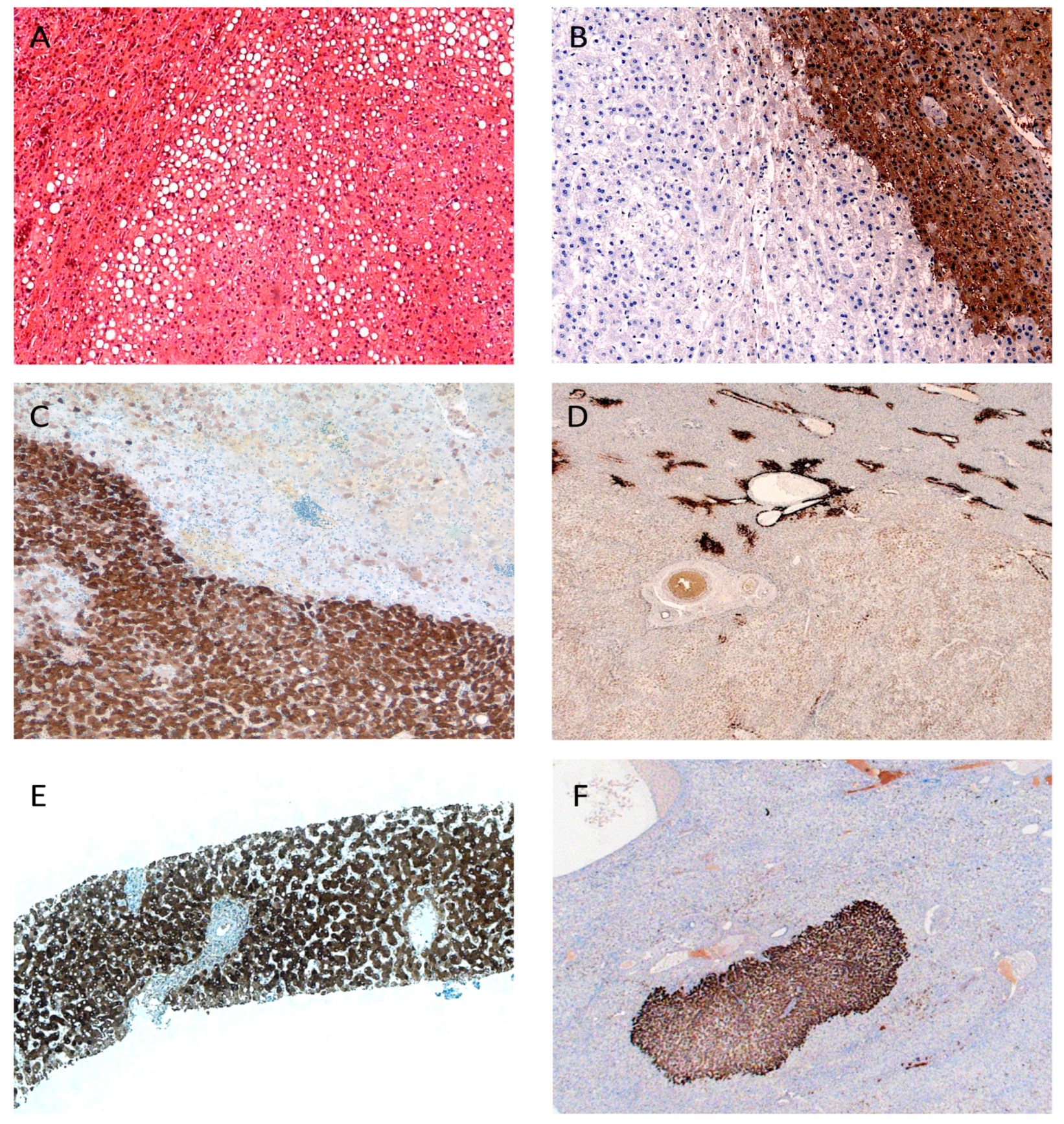
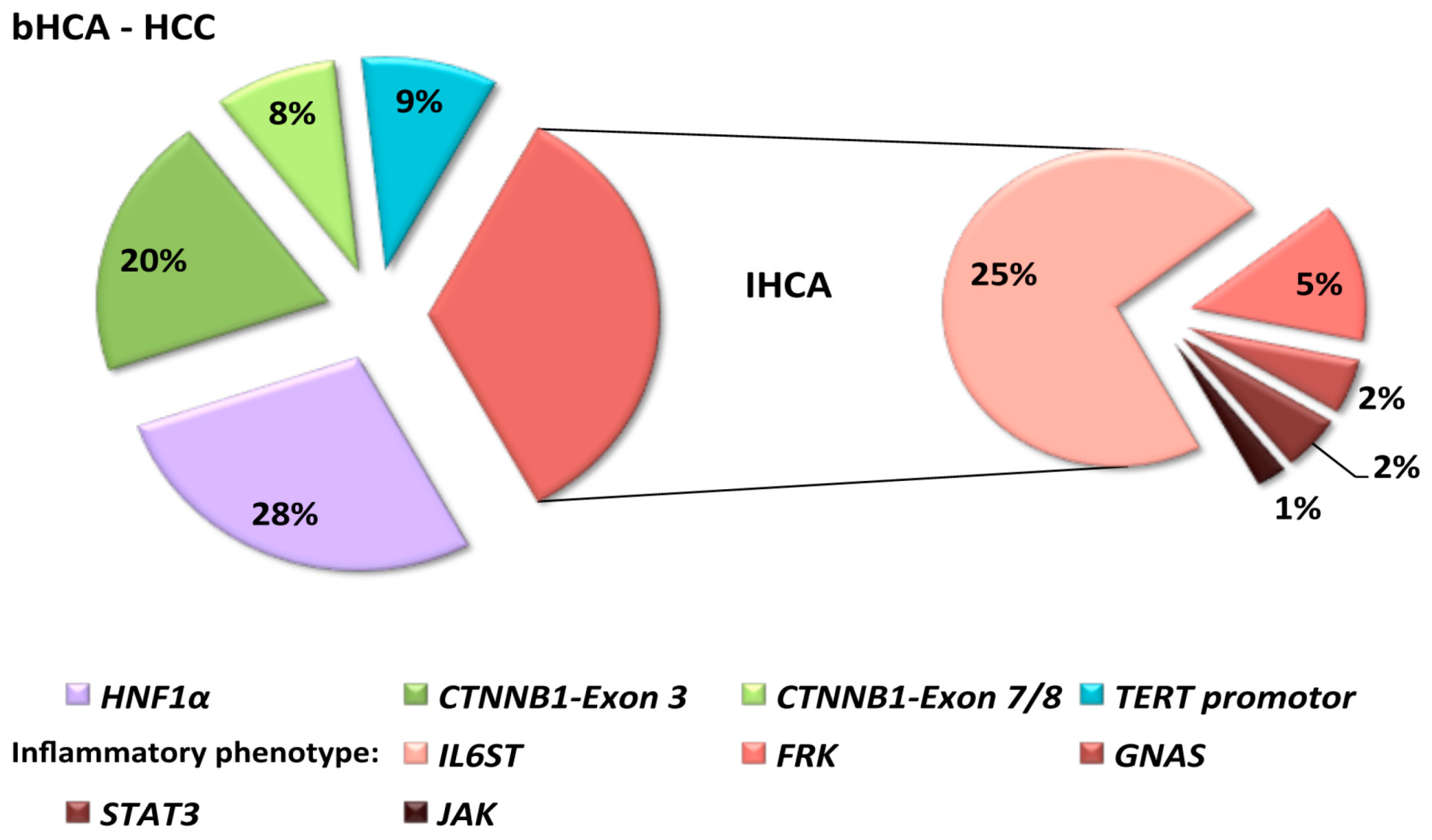
2. Hepatocytic Nuclear Factor 1 α (HNF1α) Inactivated Hepatocellular Adenomas (HCA)

3. β-Catenin Activated HCA
4. Inflammatory HCA
5. Unclassified HCA
6. Adenomatosis
7. Differential Diagnosis of HCA and Hepatocellular Carcinoma (HCC)
8. Future Perspectives
9. Conclusions

Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mettlin, C.; Natarajan, N. Studies on the role of oral contraceptive use in the etiology of benign and malignant liver tumors. J. Surg. Oncol. 1981, 18, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Labrune, P.; Morcrette, G.; Rebouissou, S.; Franco, D.; Prevot, S.; Quaglia, A.; Bedossa, P.; Libbrecht, L.; Terracciano, L.; et al. Molecular characterization of hepatocellular adenomas developed in patients with glycogen storage disease type I. J. Hepatol. 2013, 58, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Fabre, M.; Couchy, G.; Pilati, C.; Jeannot, E.; Tran Van Nhieu, J.; Saint-Paul, M.C.; de Muret, A.; Redon, M.J.; Buffet, C.; et al. GNAS-activating mutations define a rare subgroup of inflammatory liver tumors characterized by STAT3 activation. J. Hepatol. 2012, 56, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Willson, J.S.; Godwin, T.D.; Wiggins, G.A.; Guilford, P.J.; McCall, J.L. Primary hepatocellular neoplasms in a MODY3 family with a novel HNF1α germline mutation. J. Hepatol. 2013, 59, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Bioulac-Sage, P.; Laumonier, H.; Couchy, G.; Le Bail, B.; Sa Cunha, A.; Rullier, A.; Laurent, C.; Blanc, J.F.; Cubel, G.; Trillaud, H.; et al. Hepatocellular adenoma management and phenotypic classification: The Bordeaux experience. Hepatology 2009, 50, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Stoot, J.H.; Coelen, R.J.; De Jong, M.C.; Dejong, C.H. Malignant transformation of hepatocellular adenomas into hepatocellular carcinomas: A systematic review including more than 1600 adenoma cases. HPB 2010, 12, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Farges, O.; Ferreira, N.; Dokmak, S.; Belghiti, J.; Bedossa, P.; Paradis, V. Changing trends in malignant transformation of hepatocellular adenoma. Gut 2011, 60, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Bioulac-Sage, P.; Sempoux, C.; Possenti, L.; Frulio, N.; Laumonier, H.; Laurent, C.; Chiche, L.; Blanc, J.F.; Saric, J.; Trillaud, H.; et al. Pathological diagnosis of hepatocellular cellular adenoma according to the clinical context. Int. J. Hepatol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Bioulac-Sage, P.; Zucman-Rossi, J. Hepatocellular benign tumors-from molecular classification to personalized clinical care. Gastroenterology 2013, 144, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Bacq, Y.; Jacquemin, E.; Balabaud, C.; Jeannot, E.; Scotto, B.; Branchereau, S.; Laurent, C.; Bourlier, P.; Pariente, D.; de Muret, A.; et al. Familial liver adenomatosis associated with hepatocyte nuclear factor 1α inactivation. Gastroenterology 2003, 125, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- Bluteau, O.; Jeannot, E.; Bioulac-Sage, P.; Marques, J.M.; Blanc, J.F.; Bui, H.; Beaudoin, J.C.; Franco, D.; Balabaud, C.; Laurent-Puig, P.; et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat. Genet. 2002, 32, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, E.; Mellottee, L.; Bioulac-Sage, P.; Balabaud, C.; Scoazec, J.Y.; Tran Van Nhieu, J.; Bacq, Y.; Michalak, S.; Buob, D.; Groupe d’etude Genetique des Tumeurs, H.; et al. Spectrum of HNF1α somatic mutations in hepatocellular adenoma differs from that in patients with MODY3 and suggests genotoxic damage. Diabetes 2010, 59, 1836–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucman-Rossi, J.; Jeannot, E.; Nhieu, J.T.; Scoazec, J.Y.; Guettier, C.; Rebouissou, S.; Bacq, Y.; Leteurtre, E.; Paradis, V.; Michalak, S.; et al. Genotype-phenotype correlation in hepatocellular adenoma: New classification and relationship with hcc. Hepatology 2006, 43, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilati, C.; Letouze, E.; Nault, J.C.; Imbeaud, S.; Boulai, A.; Calderaro, J.; Poussin, K.; Franconi, A.; Couchy, G.; Morcrette, G.; et al. Genomic profiling of hepatocellular adenomas reveals recurrent FRK-activating mutations and the mechanisms of malignant transformation. Cancer Cell 2014, 25, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Balabaud, C.; Al-Rabih, W.R.; Chen, P.J.; Evason, K.; Ferrell, L.; Hernandez-Prera, J.C.; Huang, S.F.; Longerich, T.; Park, Y.N.; Quaglia, A.; et al. Focal nodular hyperplasia and hepatocellular adenoma around the world viewed through the scope of the immunopathological classification. Int. J. Hepatol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.; Hoton, D.; Dardenne, S.; Annet, L.; Hubert, C.; Godecharles, S.; Jouret-Mourin, A.; Reding, R.; Otte, J.B.; Rahier, J.; et al. Histological and immunohistochemical revision of hepatocellular adenomas: A learning experience. Int. J. Hepatol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.E.; Ward, J.M.; Gonzalez, F.J. Regulation of the liver fatty acid-binding protein gene by hepatocyte nuclear factor 1α (HNF1α). Alterations in fatty acid homeostasis in HNF1α-deficient mice. J. Biol. Chem. 2000, 275, 27117–27122. [Google Scholar] [PubMed]
- Odom, D.T.; Zizlsperger, N.; Gordon, D.B.; Bell, G.W.; Rinaldi, N.J.; Murray, H.L.; Volkert, T.L.; Schreiber, J.; Rolfe, P.A.; Gifford, D.K.; et al. Control of pancreas and liver gene expression by hnf transcription factors. Science 2004, 303, 1378–1381. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, L.; Rebouissou, S.; Paris, A.; Rathahao-Paris, E.; Perdu, E.; Bioulac-Sage, P.; Imbeaud, S.; Zucman-Rossi, J. Loss of HNF1α function in human hepatocellular adenomas leads to aberrant activation of signaling pathways involved in tumorigenesis. Hepatology 2010, 51, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Oda, N.; Kaisaki, P.J.; Menzel, S.; Furuta, H.; Vaxillaire, M.; Southam, L.; Cox, R.D.; Lathrop, G.M.; Boriraj, V.V.; et al. Mutations in the hepatocyte nuclear factor-1α gene in maturity-onset diabetes of the young (MODY3). Nature 1996, 384, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, E.; Poussin, K.; Chiche, L.; Bacq, Y.; Sturm, N.; Scoazec, J.Y.; Buffet, C.; van Nhieu, J.T.; Bellanne-Chantelot, C.; de Toma, C.; et al. Association of CYP1B1 germ line mutations with hepatocyte nuclear factor 1α-mutated hepatocellular adenoma. Cancer Res. 2007, 67, 2611–2616. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.D.; Liehr, J.G. Molecular mechanisms of estrogen carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 203–232. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Thorgeirsson, S.S. Next-generation genomic profiling of hepatocellular adenomas: A new era of individualized patient care. Cancer Cell 2014, 25, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Soe, K.L.; Soe, M.; Gluud, C. Liver pathology associated with the use of anabolic-androgenic steroids. Liver 1992, 12, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Sempoux, C.; Balabaud, C.; Bioulac-Sage, P. Malignant transformation of hepatocellular adenoma. Hepat. Oncol. 2014, 1, 421–431. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Jeng, Y.M.; Yeh, S.H.; Chen, P.J. P53 gene and Wnt signaling in benign neoplasms: β-catenin mutations in hepatic adenoma but not in focal nodular hyperplasia. Hepatology 2002, 36, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Levy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of β-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef] [PubMed]
- Rebouissou, S.; Couchy, G.; Libbrecht, L.; Balabaud, C.; Imbeaud, S.; Auffray, C.; Roskams, T.; Bioulac-Sage, P.; Zucman-Rossi, J. The β-catenin pathway is activated in focal nodular hyperplasia but not in cirrhotic FNH-like nodules. J. Hepatol. 2008, 49, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micsenyi, A.; Tan, X.; Sneddon, T.; Luo, J.H.; Michalopoulos, G.K.; Monga, S.P. β-catenin is temporally regulated during normal liver development. Gastroenterology 2004, 126, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balabaud, C.; Bioulac-Sage, P. Glutamine synthetase interpretation in hepatocellular adenoma. Virchows Arch. 2014, 465, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Bioulac-Sage, P.; Rebouissou, S.; Thomas, C.; Blanc, J.F.; Saric, J.; Sa Cunha, A.; Rullier, A.; Cubel, G.; Couchy, G.; Imbeaud, S.; et al. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology 2007, 46, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Fukusato, T.; Soejima, Y.; Kondo, F.; Inoue, M.; Watanabe, M.; Takahashi, Y.; Aso, T.; Uozaki, H.; Sano, K.; Sanada, Y.; et al. Preserved or enhanced OATP1b3 expression in hepatocellular adenoma subtypes with nuclear accumulation of β-catenin. Hepatol. Res. 2014, 24. [Google Scholar] [CrossRef]
- Ueno, A.; Masugi, Y.; Yamazaki, K.; Komuta, M.; Effendi, K.; Tanami, Y.; Tsujikawa, H.; Tanimoto, A.; Okuda, S.; Itano, O.; et al. OATP1b3 expression is strongly associated with Wnt/β-catenin signalling and represents the transporter of gadoxetic acid in hepatocellular carcinoma. J. Hepatol. 2014, 61, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Shafizadeh, N.; Genrich, G.; Ferrell, L.; Kakar, S. Hepatocellular adenomas in a large community population, 2000 to 2010: Reclassification per current world health organization classification and results of long-term follow-up. Hum. Pathol. 2014, 45, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Burt, A.D.; Brunt, E.M.; Callea, F.; Clouston, A.D.; Dienes, H.P.; Goodman, Z.D.; Gouw, A.S.; Hubscher, S.G.; Roberts, E.A.; et al. Well-differentiated hepatocellular neoplasm of uncertain malignant potential: Proposal for a new diagnostic category. Hum. Pathol. 2014, 45, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Balabaud, C.; Bioulac-Sage, P.; Ferrell, L.; Kakar, S.; Paradis, V.; Quaglia, A.; Sempoux, C.; Thung, S.; Zucman-Rossi, J. Well-differentiated hepatocellular neoplasm of uncertain malignant potential. Hum. Pathol. 2015, 46, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Pinyol, R.; Tovar, V.; Llovet, J.M. TERT promoter mutations: Gatekeeper and driver of hepatocellular carcinoma. J. Hepatol. 2014, 61, 685–687. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Marrero, J.A.; Fontana, R.J.; Fu, S.; Conjeevaram, H.S.; Su, G.L.; Lok, A.S. Alcohol, tobacco and obesity are synergistic risk factors for hepatocellular carcinoma. J. Hepatol. 2005, 42, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Poussin, K.; Pilati, C.; Couchy, G.; Calderaro, J.; Bioulac-Sage, P.; Bacq, Y.; Paradis, V.; Leteurtre, E.; Sturm, N.; Ramos, J.; et al. Biochemical and functional analyses of gp130 mutants unveil JAK1 as a novel therapeutic target in human inflammatory hepatocellular adenoma. Oncoimmunology 2013, 2, e27090. [Google Scholar] [CrossRef] [PubMed]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, Z.; Zheng, H.; Liu, X.; Li, S.; Barber, T.D.; Gong, Z.; Gao, H.; Hao, K.; Willard, M.D.; Xu, J.; et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013, 23, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Fabre, A.; Audet, P.; Vilgrain, V.; Nguyen, B.N.; Valla, D.; Belghiti, J.; Degott, C. Histologic scoring of liver biopsy in focal nodular hyperplasia with atypical presentation. Hepatology 2002, 35, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Tannapfel, A.; Busse, C.; Geissler, F.; Witzigmann, H.; Hauss, J.; Wittekind, C. INK4a-ARF alterations in liver cell adenoma. Gut 2002, 51, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Frulio, N.; Chiche, L.; Bioulac-Sage, P.; Balabaud, C. Hepatocellular adenomatosis: What should the term stand for! Clin. Res. Hepatol. Gastroenterol. 2014, 38, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Castain, C.; Sempoux, C.; Brunt, E.M.; Causse, O.; Heitzmann, A.; Hernandez-Prera, J.C.; le Bail, B.; Schirmacher, P.; Thung, S.N.; Balabaud, C.; et al. Coexistence of inflammatory hepatocellular adenomas with HNF1α-inactivated adenomas: Is there an association? Histopathology 2014, 64, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Rake, J.P.; Visser, G.; Labrune, P.; Leonard, J.V.; Ullrich, K.; Smit, G.P. Glycogen storage disease type I: Diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur. J. Pediatr. 2002, 161, S20–S34. [Google Scholar] [CrossRef] [PubMed]
- Labrune, P.; Trioche, P.; Duvaltier, I.; Chevalier, P.; Odievre, M. Hepatocellular adenomas in glycogen storage disease type I and III: A series of 43 patients and review of the literature. J. Pediatr. Gastroenterol. Nutr. 1997, 24, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Sakagami, K.; Nakata, Y.; Komazawa, K.; Amimoto, T.; Nakashima, K.; Isozaki, H.; Takakura, N.; Tanaka, N. Multiple hepatic adenomas caused by long-term administration of androgenic steroids for aplastic anemia in association with familial adenomatous polyposis. J. Gastroenterol. 2000, 35, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.M.; Petersen, G.M.; Krush, A.J.; Booker, S.; Jen, J.; Giardiello, F.M.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Molecular diagnosis of familial adenomatous polyposis. N. Engl. J. Med. 1993, 329, 1982–1987. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Wunsch, P.H.; Ballhausen, W.G. Childhood hepatocellular adenoma in familial adenomatous polyposis: Mutations in adenomatous polyposis coli gene and p53. Gastroenterology 1997, 112, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Sheridan, R.M.; Towbin, A.; Geller, J.I.; Tiao, G.; Bove, K.E. Multifocal hepatic neoplasia in 3 children with APC gene mutation. Am. J. Surg. Pathol. 2013, 37, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Blaker, H.; Sutter, C.; Kadmon, M.; Otto, H.F.; Von Knebel-Doeberitz, M.; Gebert, J.; Helmke, B.M. Analysis of somatic APC mutations in rare extracolonic tumors of patients with familial adenomatous polyposis coli. Genes Chromosomes Cancer 2004, 41, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Sakaguchi, T.; Kurachi, K.; Mori, H.; Tao, H.; Nakamura, T.; Takehara, Y.; Baba, S.; Maekawa, M.; Sugimura, H.; et al. Hepatocellular adenoma associated with familial adenomatous polyposis coli. World J. Hepatol. 2012, 4, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, E.; Wendum, D.; Paye, F.; Mourra, N.; de Toma, C.; Flejou, J.F.; Zucman-Rossi, J. Hepatocellular adenoma displaying a HNF1α inactivation in a patient with familial adenomatous polyposis coli. J. Hepatol. 2006, 45, 883–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toiyama, Y.; Inoue, Y.; Yasuda, H.; Yoshiyama, S.; Araki, T.; Miki, C.; Kusunoki, M. Hepatocellular adenoma containing hepatocellular carcinoma in a male patient with familial adenomatous polyposis coli: Report of a case. Surg. Today 2011, 41, 1442–1446. [Google Scholar] [CrossRef] [PubMed]
- Coston, W.M.; Loera, S.; Lau, S.K.; Ishizawa, S.; Jiang, Z.; Wu, C.L.; Yen, Y.; Weiss, L.M.; Chu, P.G. Distinction of hepatocellular carcinoma from benign hepatic mimickers using glypican-3 and CD34 immunohistochemistry. Am. J. Surg. Pathol. 2008, 32, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Lagana, S.M.; Salomao, M.; Bao, F.; Moreira, R.K.; Lefkowitch, J.H.; Remotti, H.E. Utility of an immunohistochemical panel consisting of glypican-3, heat-shock protein-70, and glutamine synthetase in the distinction of low-grade hepatocellular carcinoma from hepatocellular adenoma. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Kim, C.J.; Jeon, E.J.; Sung, C.O.; Shin, H.J.; Choi, J.; Yu, E. Overexpression of C-reactive protein as a poor prognostic marker of resectable hepatocellular carcinomas. J. Pathol. Transl. Med. 2015, 49, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Takahashi, Y.; Fujii, T.; Kitagawa, M.; Fukusato, T. Significance of downregulation of liver fatty acid-binding protein in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 17541–17551. [Google Scholar] [CrossRef] [PubMed]
- Bioulac-Sage, P.; Laumonier, H.; Cubel, G.; Rossi, J.Z.; Balabaud, C. Hepatic resection for inflammatory hepatocellular adenomas: Pathological identification of micronodules expressing inflammatory proteins. Liver Int. 2010, 30, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Forner, A.; Korangy, F.; N’Kontchou, G.; Barget, N.; Ayuso, C.; Ormandy, L.A.; Manns, M.P.; Beaugrand, M.; Bruix, J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goltz, D.; Fischer, H.-P. Current Proceedings in the Molecular Dissection of Hepatocellular Adenomas: Review and Hands-on Guide for Diagnosis. Int. J. Mol. Sci. 2015, 16, 20994-21007. https://doi.org/10.3390/ijms160920994
Goltz D, Fischer H-P. Current Proceedings in the Molecular Dissection of Hepatocellular Adenomas: Review and Hands-on Guide for Diagnosis. International Journal of Molecular Sciences. 2015; 16(9):20994-21007. https://doi.org/10.3390/ijms160920994
Chicago/Turabian StyleGoltz, Diane, and Hans-Peter Fischer. 2015. "Current Proceedings in the Molecular Dissection of Hepatocellular Adenomas: Review and Hands-on Guide for Diagnosis" International Journal of Molecular Sciences 16, no. 9: 20994-21007. https://doi.org/10.3390/ijms160920994
APA StyleGoltz, D., & Fischer, H.-P. (2015). Current Proceedings in the Molecular Dissection of Hepatocellular Adenomas: Review and Hands-on Guide for Diagnosis. International Journal of Molecular Sciences, 16(9), 20994-21007. https://doi.org/10.3390/ijms160920994