1. Introduction
Nodal ligands are potent morphogens, belonging to the TGF-β superfamily [
1,
2,
3,
4]. In humans, Nodal is rarely expressed during late development and adulthood. Nodal physiological expression is typically observed only in embryonic tissues and embryonic stem (ES) cells. Aberrant re-activation of Nodal expression in adults is associated with a number of tumors such as metastatic melanoma as well as breast, colon, prostate, and ovarian carcinomas [
5,
6,
7,
8].
During the last years, increased attention has been devoted to the correlation existing between the expression levels of Nodal and melanoma development. These studies have associated the hyper-activation of Nodal signalling to melanoma invasiveness, aggressiveness, plasticity, and tumorigenicity [
5,
9,
10,
11,
12,
13].
In human metastatic melanoma cells, the up-regulation of Nodal is controlled by the epigenetic silencing of the antagonist Lefty [
10,
14,
15] and by a positive cross-talk with the Notch4 ligand [
16,
17].
Under physiological conditions, Nodal signalling plays a crucial role in early embryonic and neural development and in the maintaining of pluripotency in ES cells. It is synthesized by cells as pre-pro-Nodal (55 kDa) and subsequently converted to the intermediate pro-Nodal (37 kDa) and finally to the mature and active Nodal (22 kDa) by a proteolytical process induced by subtilisin-like proprotein convertases PACE-4 and Furin. Once secreted from cells, Nodal can act as either autocrine or paracrine agent.
Nodal signalling is triggered by interaction with heteromultimeric complexes consisting of the EGF-like domain of the co-receptor Cripto-1 and type I (ALK 4/7) and type II (ActRIIB) activin-like serine-threonine kinase receptors. Such association leads to the phosphorylation and activation of ALK 4/7 by ActRIIB which, in turn, promotes the phosphorylation of Smad2/3. Hence, phosphorylated Smad-2/3 proteins associate with the common mediator Smad-4 prior to translocation to the nucleus where, together with transcription factors such as FoxH1, Mixer, and p53, regulate expression of some target genes. These latter include Nodal as well as Lefty A and B, which are natural extracellular inhibitors. Interestingly, Nodal activates its own expression via a positive feedback loop.
Nodal signalling can occur in the extracellular microenvironment, where Nodal and Cripto-1 are present as free ligands, as well as on the cellular membrane surface, in the form of a multimeric complex. It is well established that Cripto-1 is essential for the cellular activation of Nodal/ALK4/Smad pathway [
18] and that it may directly interact with ALK4 to enhance the responsiveness of the ALK7/ActRIIB complex to Nodal [
19]. Recently we have reported that Nodal is also capable of physically interacting with both ALK7 and ALK4 regardless of the Cripto-1 presence [
20,
21]. Although the exact mechanisms underlying the synergistic effect exerted by Cripto-1 on the cellular activation of Nodal-dependent Smad signalling have not been completely understood, the detection of Cripto-1 in melanoma cells [
5,
22,
23] supports the relevant role this co-receptor can play in co-promoting tumorigenesis.
Like other ligands of the TGF-β superfamily, Nodal can activate additional signalling pathways such as MAPK/ERK (ras/raf/mitogen-activated protein kinase) and PI3K–AKT. MAPK signalling, which converges on ERK and is related to the single mutation in the BRAF protein, has been also reported as a relevant pathway involved in melanoma disease and progression [
24,
25].
These findings suggest that the quantification of Nodal expression in cancer tissues is both a diagnostic and prognostic option and designate the protein as a biomarker and as a potential new therapeutic target in melanoma and other tumors.
Multiple pharmacological therapies have been approved for malignant melanoma, including BRAF inhibitors (vemurafenib and dabrafenib), MEK inhibitors (trametinib), mAbs, such as Ipilimumab (mAb anti-CTLA4) and Pembrolizumab (mAb anti-PD1), and the oldest chemotherapy drug (dacarbazine). However, many of these show a limited efficacy due to the onset of drug resistance mechanisms or their low specificity [
25,
26,
27,
28,
29,
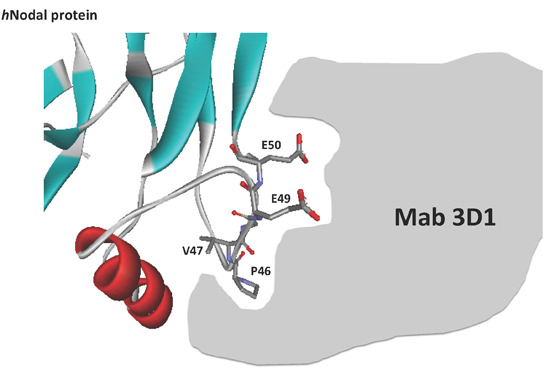
30]. The identification of more effective anti-tumor agents against novel targets, to be employed alone or in combination with those already available, remains an interesting and challenging approach. In this context, targeting and inhibition of Nodal signalling represents an attractive and alternative strategy to block melanoma progression and potentially of other cancers. With this aim, we have generated monoclonal antibodies against the Nodal 44–67 region which has been recommended as a major site of Cripto-1 recognition and as one of the key structural elements of the Nodal-Cripto-ALK4 interactions [
21,
31]. Using a subtractive ELISA screening we have selected a monoclonal antibody that recognizing the Nodal E49 and E50 hot-spot residues, and neutralizes
in vitro the binding to Cripto-1 in a specific way. We report here a full biochemical characterization of the antibody and of its functional fragments, together with preliminary data showing the selective detection of the endogenous protein in a set of human melanoma cells. Given the potential to selectively inhibit the Nodal-Cripto-Smads axis, this antibody could represent a distinctive option to block cancer progression in Nodal-positive tumor tissues [
32].
3. Experimental Section
3.1. Peptide Synthesis, Purification, and Identification
The wild-type
hNodal(44–67) peptide, its mutated variants and human GDF peptides 5, and 6/7, were prepared by step-wise solid phase synthesis as C-terminally amidated variants following standard Fmoc chemistry using an automatic SYRO system (MultiSynTech, Witten, Germany). A Rink-amide MBHA resin (Novabiochem, Laufelfingen, Switzerland) with a substitution grade of 0.57 mmol/g and amino acid derivatives with standard protections were used in all syntheses. Polypeptides were assembled under canonical conditions of peptide synthesis, using for each coupling reaction HATU/DIEA pre-activation and a five-fold excess of Fmoc-protected amino acids. Coupling and deprotection times were kept at 30 and 20 min, respectively. Standard side-chain protection groups for Fmoc chemistry were used for all residues [
38]. N-terminal acetylation was performed on the resin using acetic anhydride at 0.5 M in DMF with 5% DIEA, 20 min at room temperature. The cleavage of peptides from the solid support was performed by treatment with a trifluoroacetic acid (TFA)/tri-isopropylsilane (TIS)/water (90/5/5,
v/
v/
v) mixture for 90 min at room temperature. Crude peptides were precipitated in cold di-ethyl-ether, dissolved in a water/acetonitrile (1/1,
v/
v) mixture and lyophilized. After lyophilisation, peptides were dissolved in a solution of H
2O/CH
3CN 95/5
v/
v, containing 0.1% TFA. They were then purified by reverse-phase HPLC (RP-HPLC) on a WATERS Prep 150 LC preparative system (Waters, Milan, Italy) using a semi-preparative 10 × 1 cm ID C18 monolythic Onyx column (Phenomenex, Torrance, CA, USA), applying a linear gradient of 0.05% TFA in CH
3CN from 5% to 70% over 10 min at a flow rate of 15 mL/min, and monitoring the absorbance at 214 nm. The collected fractions were lyophilized. Peptide purity and identity were confirmed by liquid chromatography–mass spectrometry analysis (LC–MS); analyses were carried out on a LCQ DECA XP ion Trap mass spectrometer (ThermoFisher, Waltham, MA, USA) equipped with an OPTON ESI source and with a complete Surveyor HPLC system. Typical gradients applied to elute the peptides were as follows: flow rate 0.2 mL/min; gradients from 5% solvent B (ACN, 0.05% TFA) to 60% solvent B in 10 min. Solvent A was H
2O, 0.08% TFA. Biobasic C18 50 × 2 mm ID columns (ThermoFisher, Milano, Italy) were used to separate peptides during LC–MS analyses.
3.2. Immunogen Preparation
One mg of non-acetylated
hNodal(44–67) peptide was conjugated with 3.0 mg carrier proteins (KLH and BSA) in 2.0 mL of 20 mM phosphate buffer pH 7.0 containing 0.2%
v/
v glutaraldehyde (stock solution 25%), by stirring the mixture for 3 h at room temperature [
39]. The reaction was blocked by adding 1.0 mL of 1.0 M glycine in water, then solutions were extensively dialyzed against PBS buffer pH 7.4 before being lyophilized. The amount of peptide-protein conjugate was determined using the Bradford assay [
40].
3.3. Antibody Generation
BALB/c mice were housed and handled according to the institutional guidelines (Project identification code 2013/0038120, approved by the Ethical Animal Care and Use Committee, University of Naples “Federico II”. Date of approval 24 April 2013). Four five-week old female BALB/c mice (Jackson Lab) were immunized by sub cutaneous injection with 300 μL of suspension containing 100 μg of KLH-conjugated
hNodal(44–67) peptide mixture emulsified with Complete Freund’s Adjuvant. Before immunization, 250 µL blood samples were taken from each mouse from the caudal vein and used as the pre-immune control (T0 samples). Mice were boosted with the same amount of immunogen in incomplete Freund’s adjuvant at day 18 and at day 30; blood samples were taken from the caudal vein (250 µL) before every subsequent immunization and tested by ELISA to monitor antibody titer. A final antigen boost was administered sub-cutis in to the mice showing the highest antibody titer seven days before being sacrificed and splenectomised as described below. Cells harvested from spleens of sacrificed animals were fused with myeloma SP2/0 (ATCC) cells at a ratio of 5:1 in RPMI-GM containing polyethylene glycol (PEG) 1300–1600 (Hybri-Max, Sigma–Aldrich, Milano, Italy) and 7.5% DMSO (Sigma–Aldrich, Milano, Italy) as described by Köhler [
41].
The fused hybridoma cells were re-suspended in 30 mL of selection medium consisting of RPMI–GM medium containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 100 μM hypoxanthine, 16 μM thymidine and 400 nM aminopterin (RPMI-HAT Sigma–Aldrich, Milano, Italy). The cell suspension (200 μL) was dispensed into 96-well plates and incubated at 37 °C in a 5% CO
2 atmosphere. Between day 12 and 14, supernatants were screened by ELISA for binding to
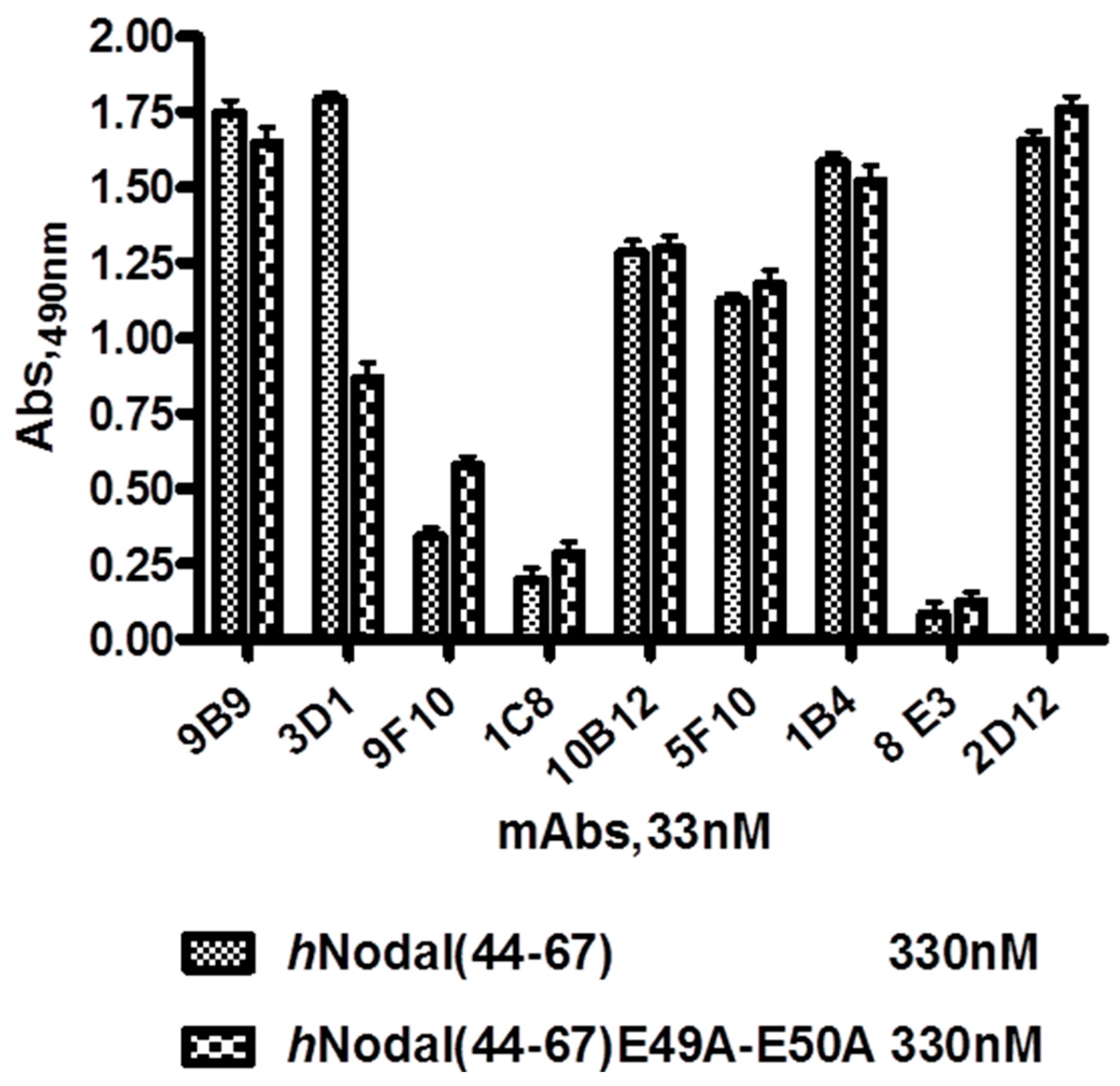
hNodal(44–67) peptide and its mutated variant
hNodal(44–67)E49A–E50A, in which the glutamic acids 49 and 50 were substituted with two alanine residues. A detailed description of the ELISA procedure is given in the on-line
Supplementary Material.
The hybridoma clone named 3D1, with strong reactivity with hNodal(44–67) peptide (but not with the mutated variant) was re-cloned twice by limited dilution and its reactivity was re-confirmed by ELISA. Sub-cloned hybridoma cells were cultured in RPMI-HAT containing 10% FBS and slowly adopted to serum-free medium. The OPTI-MEM medium containing 10% FBS. Adapted cells were cultured in Optimem medium and then transferred to the Bioreactor (Bio Cell Line, Becton Dickinson, Franklin Lakes, NJ, USA) for large scale production.
Isotype of the anti-Nodal antibodies produced by the selected clones was determined by using the commercial kit specific for murine antibodies, in accordance to the manufacturer’s instructions (Pierce Rapid Mouse antibody Isotyping kit, Thermo Scientific, Waltham, MA, USA).
3.4. ELISA: Antibody Titration in Mouse Sera
To determine the antibody titer in mouse sera, 96-well ELISA plates were coated with 50 µL of the BSA-conjugated hNodal(44–67), 1 μg/mL in PBS, overnight at 4 °C. The plate was then washed three times with PBS containing 0.05% Tween 20 (PBS-T). Non-specific sites of the plate were blocked with 5% BSA and incubated at R.T. for 120 min. Wells were then washed three times with PBS-T and serum was (50 μL) added to the wells in three-fold serial dilutions starting from 1:100 to 1:100,000. The plate was incubated at 37 °C for 1 h and washed again with PBS-T. Then, 50 μL of a 1:1000 dilution of HRP-conjugated rabbit anti-mouse Ig (1.0 mg/mL, Biorad, Milano, Italy) were added to the wells and incubation was carried out for 1 h at 37 °C. After washing, 100 μL of 2,2-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) substrate solution was added to each well. After 15 min, the reaction was stopped by adding 50 μL of 1% SDS solution in water to each well. The Optical Density (OD) was measured at 415 nm by a microplate reader (BioTek, Winooski, VT, USA). Antibody titers were evaluated sufficiently high when the average absorbance values from triplicate wells incubated with immune sera dilutions at 1:10,000 were at least thrice those determined on wells incubated with the pre-immune serum.
3.5. ELISA: Screening of Hybridoma Supernatants
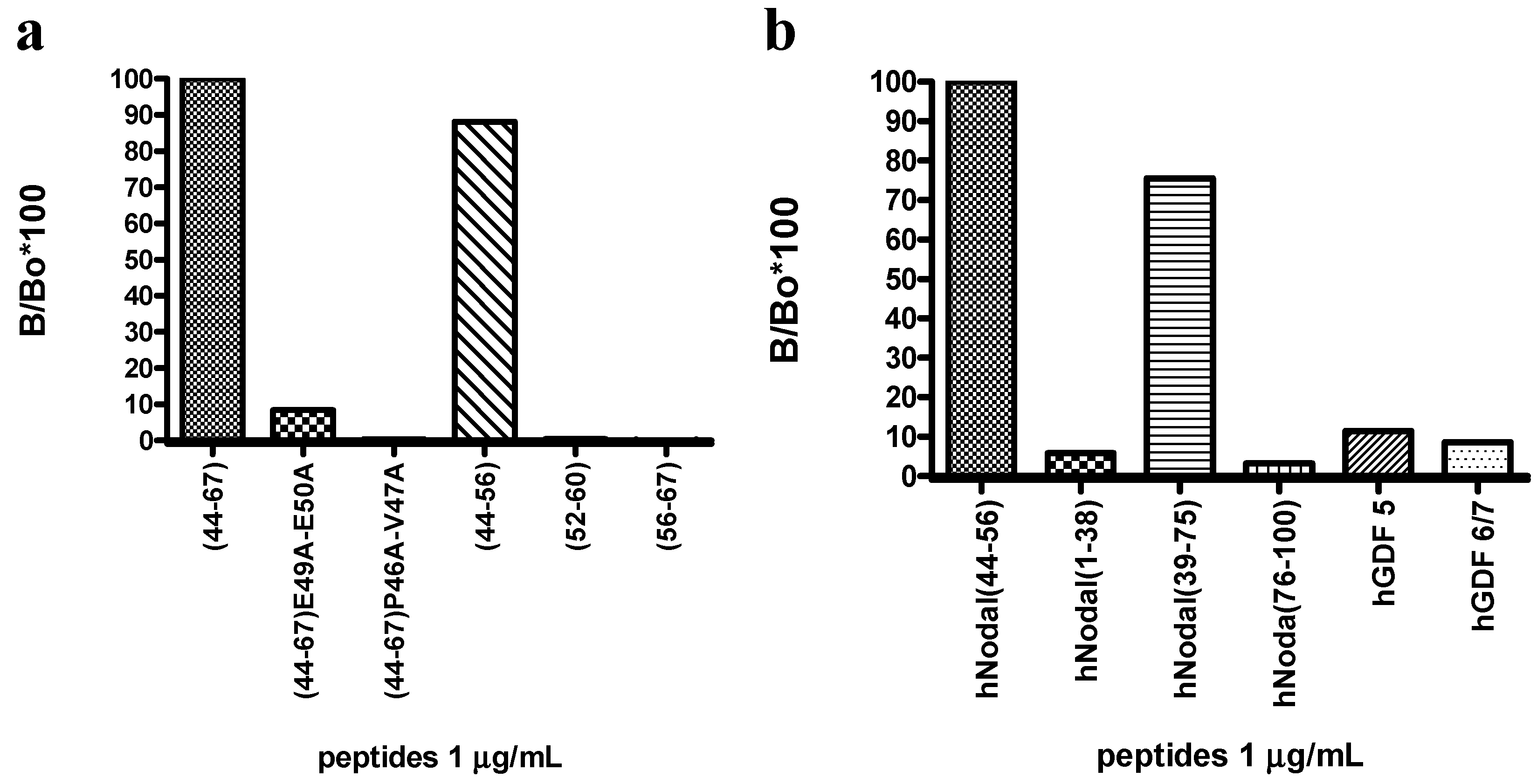
For hybridoma supernatants screening, hNodal(44–67) and hNodal(44–67)E49A–E50A were coated at 330 nM diluted in PBS on polystyrene conical flat bottom 96-well plates (Nunc MaxiSorp™, Roskilde, Denmark) at 4 °C overnight (100 µL/well). After incubation, the coated wells were washed three times with PBS containing 0.005% Tween-20 (PBS-T) and non-specific binding sites were blocked by incubating with 1% BSA in PBS (300 µL/well) for 1 h at 37 °C. After washing three times with PBS-T, supernatants of hybridomas diluted in PBS at 33 nM were incubated for 1 h at 37 °C (100 µL/well). After incubation, plates were triple washed with PBS-T and goat anti-mouse HRP-conjugated antibody (1 mg/mL, Blotting Grade Affinity Purified Goat Anti-Mouse IgG (H + L)) diluted 1:1000 in PBS was added as secondary antibody (100 µL/well) and incubated for 1 h at 37 °C. After incubation and three washes with PBS-T, bound antibodies were detected by adding 100 µL/well of freshly prepared O-phenylenediamine (0.4 mg/mL) containing H2O2 (0.4 mg/mL) in 0.1 M citrate buffer (pH 5.2) (SIGMAFAST™ OPD tablet Sigma–Aldrich). The peroxidase reaction was stopped after 5 min with 50 µL/well of 2.5 M H2SO4 and the optical density was measured at 490 nm, using a microplate reader. The dose-dependent binding of 3D1 to immobilized hNodal(44–67) and hNodal(44–67)E49A–E50A was performed by coating the peptides at 60 nM and using 3D1 at concentrations between 0.1 and 66 nM.
3.6. ELISA: Epitope Mapping and Specificity Assay
Peptides used for the epitope mapping and for the specificity assay of the 3D1 mAb were coated at 1.0 µg/mL diluted in PBS; 3D1 mAb, dissolved in PBS, was used as primary antibody at 1.0 µg/mL. The ELISA assays were carried out following the already described procedure.
3.7. Purification of Monoclonal Antibodies and F(ab′)2/Fab′ Fragments
Antibodies and F(ab′)
2/Fab′ fragments were purified by standard procedures. After centrifugation at 4000 rpm for 20 min at 4 °C, hybridoma cell-culture supernatants, containing monoclonal IgGs, were filtered on a 0.22 μm filter and loaded onto a HiTrap™ Protein G HP column (GE Healthcare). Purifications were performed on an ÄKTA FPLC™ instrument (GE Healthcare) at a flow rate of 0.6 mL/min, monitoring the absorbance at 280 nm. After washing away the unbound material using PBS (pH 7.4) as loading buffer, bound antibodies were recovered by changing drastically the pH conditions using 100 mM Glycine pH 2.7 as elution buffer. Eluted antibodies were quickly neutralized by adding 1/10 volume 2 M TRIS pH 9.0. To ensure the stability of purified mAbs and of related fragments they were buffer-exchanged into PBS (pH 7.4) and concentrated using an appropriate centrifugal filter (Millipore, Darmstadt, Germany). The concentration was estimated by using the Bradford assay [
41]. Purity of concentrated proteins were evaluated by SDS-PAGE and Coomassie blue staining.
Size-exclusion chromatography purifications were performed on a Superdex™ 200 HR 10/300 (GE Healthcare, Piscataway, NJ, USA) gel filtration column, using PBS pH 7.4 as running buffer, at a constant flow rate of 0.5 mL/min, with an elution volume of 25 mL and monitoring the absorbance at 280 nm.
3.8. SPR Analyses
All SPR analyses were performed on a Biacore 3000 instrument from GE Healthcare, using CM5 sensor chips and certified HBS buffer (20 mM HEPES, 0.15 M NaCl, pH 7.4, P20, 0.005%). Protein immobilization was carried out following the canonical amine coupling chemistry using the surface immobilization wizard procedure, operating at 5 µL/min [
42]. Channels were activated with EDC/NHS for 7 min; for the binding assays of purified mAbs to Nodal protein,
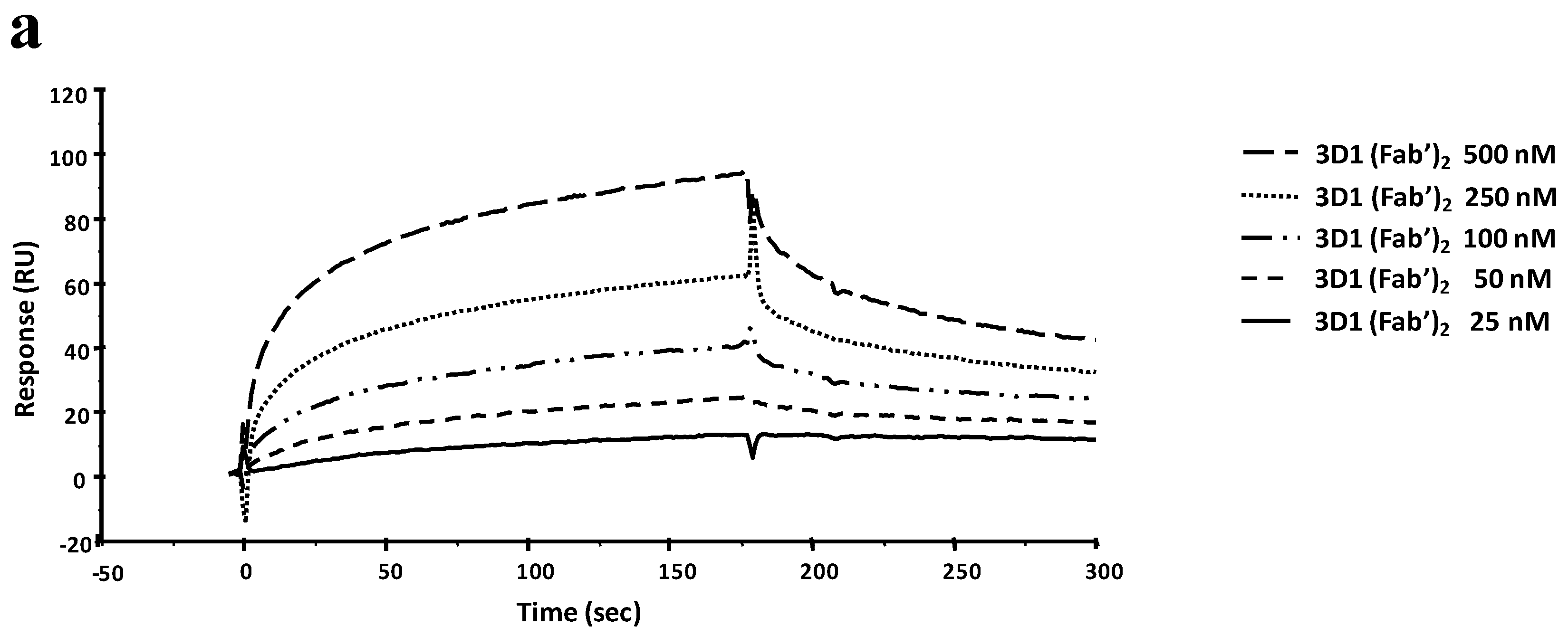
rhNodal, opportunely diluted in the pre-selected sodium acetate buffer pH 4.5, was coupled until a 4000 RU level was achieved. Residual reactive groups on the sensor chip surface were deactivated by addition of 1.0 M ethanolamine hydrochloride, pH 8.5. Antibody binding was tested at 20 µL/min injecting solutions of 3D1 (60 µL) in HBS-EP at increasing concentrations (6–100 nM). A 10 mM NaOH solution was used to regenerate the chip surface.
After the single-dose screening, dose-dependent binding analyses were carried out with the two antibodies, 3D1 and 5F10, able to bind the full-length protein. Binding of 3D1 was carried out at concentrations ranging between 6 and 100 nM. For 5F10 higher concentrations were required (100, 250, 500 and 750 nM). Dissociations were monitored for at least 500 s. To carry out the mapping of the 3D1 epitope, the whole antibody was immobilized at 5 µg/mL in 10 mM NaAc buffer pH 5 at a flow rate of 5 µL/min by standard amine coupling chemistry on a CM5 sensor chip. The immobilization level reached with the mAb was about 5350 RU. On another channel of the same sensor chip, the 3D1 Fab′ was immobilized at 10 µg/mL in 10 mM NaAc buffer pH 4.5 at a flow rate of 5 µL/min, following the standard amine coupling chemistry. The amount of immobilized Fab′ was about 2570 RU. Residual reactive groups were deactivated by 1M ethanolamine hydrochloride, pH 8.5. The blank channel was prepared as already described. Real-time binding analyses were performed at a flow rate of 20 µL/min, using HBS-EP buffer, injecting a constant sample volume of 60 µL. Peptides opportunely diluted in HBS-EP buffer were injected at the following concentrations: 0.5, 1, 2.5, 5, 10, and 20 µM. Dissociations were monitored for at least 500 s; the regeneration solution was 5 mM NaOH.
For every single analysis, experimental sensorgrams were aligned, subtracted of blank signals and overlapped. All mathematical manipulations and fitting were performed using the BiaEvaluation software, vers. 4.1 from GE Healthcare. All experimental data gave optimal fittings when processed assuming a 1:1 Langmuir binding interaction.
3.9. Antibody Deglycosylation and Pepsinolysis
To improve the splitting of the antibody by pepsin to get functional fragments, 3D1 was first subjected to a deglycosylation reaction with the Peptide-N-glycosidase F (PNGase F), supplied by New England Biolabs (Beverly, MA, USA). Following a first preliminary small-scale attempt, the large-scale deglycosylation reaction was performed incubating 1.5 mg of the mAb, diluted in PBS (pH 7.4), with 11 units of PNGase F at 37 °C. The time for optimal deglycosylation was identified as being 48 h.
Pepsinolysis reaction, monitored by SDS-PAGE, was optimized in 20 mM sodium acetate buffer, pH 4.0, using a final w/w ratio of pepsin (Sigma–Aldrich, Milano, Italy) to antibody 1:25 and incubating the mixture in a 37 °C water bath for 4 h.
3.10. Preparation of Fab′ Fragments
Fab′ fragments were produced by reducing selectively the hinge-region disulfide bonds of F(ab′)2 using 5 mM 2-Mercaptoethylamine (Thermo Scientific Pierce, Milano, Italy). Twenty mM sodium acetate buffer pH 4.0 was added to the F(ab′)2 fragments in PBS pH 7.4 to adjust the pH at 6.0 and 2 mM EDTA was also added. The mixture was incubated for 3 h at 37 °C. After incubation, PBS was added to the mixture to adjust the pH to neutrality. Reduction of F(ab′)2 to Fab′ fragments was checked and confirmed by 12% SDS-PAGE gel under non-reducing conditions. After reduction, Fab′ fragments were incubated with 25 mM IAM (Iodoacetamide) for 30 min at room temperature in the dark to block reactive thiols.
3.11. LC–ESI-TOFMS Analysis of 3D1 Fab′
The sample analyzed by mass spectrometry was reduced using 20 mM DTT for 1 h at 37 °C. Mass spectrometry analyses were performed on an Agilent 1290 Infinity LC System coupled to an Agilent 6230 time-of-flight (TOF) LC/MS System. The liquid chromatography Agilent 1290LC module was coupled with a photodiode array (PDA) detector and a 6230 time-of-flight MS detector, along with a binary solvent pump degasser, column heater, and autosampler. Based on manufacturer recommendations, the pump was connected to a gradient binary solvent system: solvent A, 0.01% TFA in H2O (v/v) and solvent B, 0.01% TFA in CH3CN (v/v). Chromatographic analyses were performed using a Phenomenex AERIS WIDEPORE reverse phase C4 3.6 mm (50 × 2.1 mm) applying a linear gradient from 25% to 65% solvent B in 15 min. The column flow rate was kept at 0.2 mL/min with the heater at a constant 20 °C. UV spectra were monitored in the range 200–400 nm. Injection volume was 1 μL followed by needle wash. The mass analyzer Agilent 6230 TOF MS was set to operate in positive ion scan mode with mass scanning from 100 to 3200 m/z. The ion source was upgraded from the original Agilent Jet Stream (AJS) source to the dual-sprayer version for improved reference mass delivery. Nitrogen was used as the drying and nebulizer gas. The instrument acquired data using the following parameters: drying gas temperature, 325 °C; drying gas flow, 10 L/min; nebulizer, 20 psi; sheath gas temperature, 400 °C; sheath gas flow, 11 L/min; VCap. 3.500 V; nozzle, 0 V; fragmentor, 200 V; skimmer, 65 V and octapole RF Vpp was 750. The instrument state was set to extended dynamic range mode (2 GHz). Tuning and calibration were performed before sample runs. Data collection and integration were performed using MassHunter workstation software (version B.05.00). Data were stored in both centroid and profile formats during acquisition. A constant flow of Agilent TOF reference solution through the reference nebulizer allowed the system to continuously correct for any mass drift by using two independent reference lock-mass ions, purine (m/z 119.03632) and HP-922 (m/z 922.000725), to ensure mass accuracy and reproducibility. Target compounds were detected and reported from accurate-mass scan data using Agilent MassHunter Qualitative software.
3.12. Competition between 3D1 Antigen Peptide and Endogenous Nodal Protein
Thirty micrograms of H9 and H14 human embryonic stem cell total protein lysates, obtained as previously described [
17,
43], were separated on a 15% SDS-PAGE gel under reducing conditions and the resolved proteins were transferred onto a PVDF membrane. Fifty ng of
rhNodal protein were used as positive control. The western blotting procedure was carried out as previously described. Membranes were blocked, then probed with the 3D1 alone at 4.0 µg/mL in 2.5% NFDM in TBS-T and with the 3D1 mAb at 4.0 µg/mL pre-incubated with its antigen, the
hNodal(44–56) peptide at 10 µg/mL. Detection was achieved with GAM-HRP (Bio-Rad) 1:1000 (1.0 µg/mL). Blots were developed with Enhanced Chemiluminescence Western Blot Substrate (Pierce, Rockford, IL, USA) following the manufacturer’s instructions and were acquired by using a Chemidoc XRS video densitometer (Bio-Rad, Hercules, CA, USA).
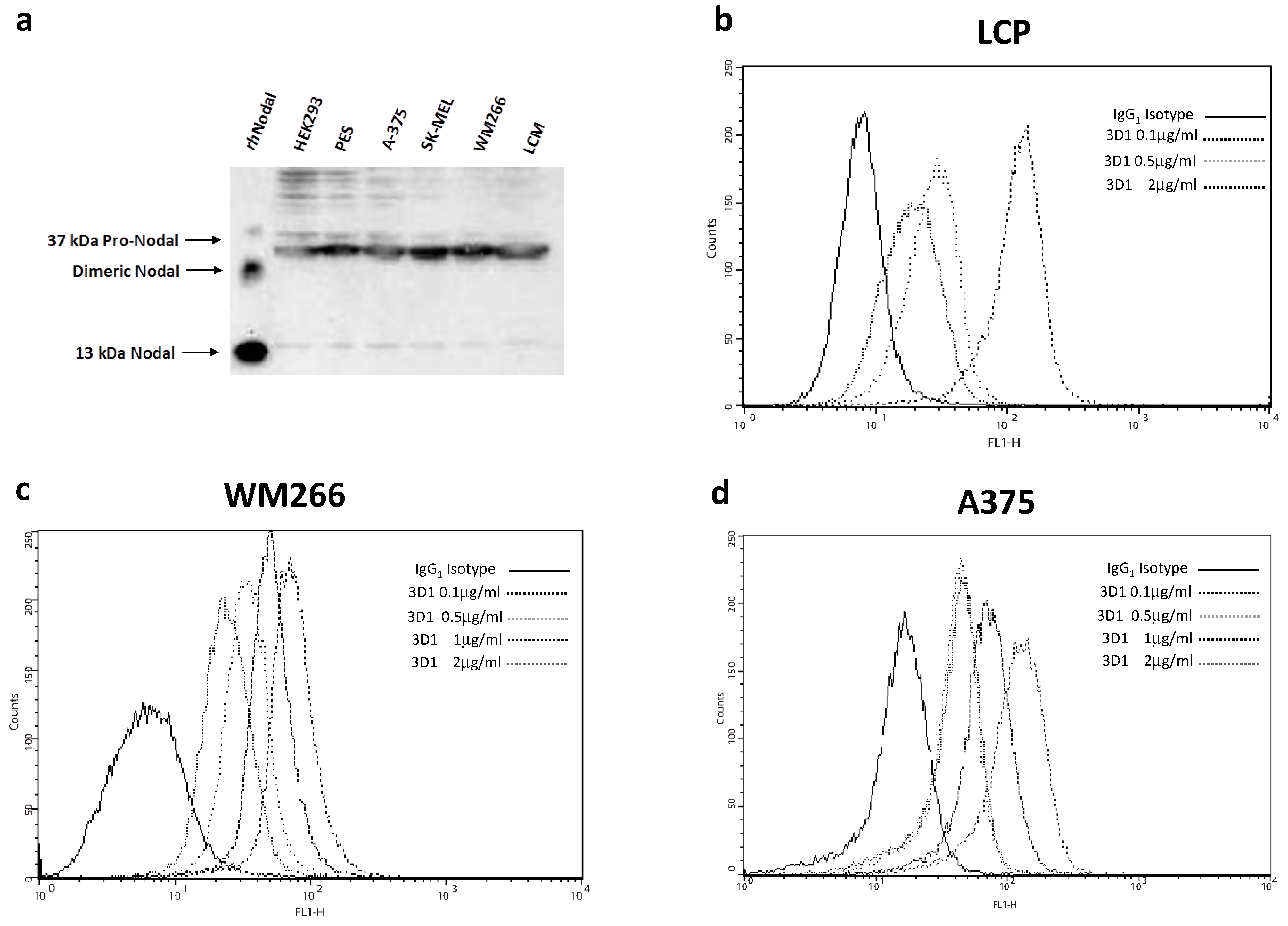
3.13. Detection of Endogenous Human Nodal in Melanoma Cells
Western blot analyses for the detection of endogenous Nodal protein were performed using the following human skin malignant melanoma cell lines: PES, A-375, SK-MEL, WM 266 and LCM. HEK-293 (human embryonic kidney) cells and 100 ng of rhNodal protein were used as positive controls. Cells were lysed performing three freeze/thaw cycles in 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 1% NP-40, 10 mM EDTA, 1 mM MgCl2, 1 mM CaCl2, 10% Glycerol, with an added cocktail of protease inhibitors (Roche, city, Switzerland) and PMSF (phenyl-methanesulfonylfluoride). Cell lysates were cleared by centrifugation at 12,000 rpm for 20 min at 4 °C and the supernatants were quantified by Bradford assay. Fifty µg of cell lysate from each sample were applied to a 15% SDS-PAGE gel under reducing conditions and electrophoresed for 3 h at 80 V. Separated proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore), previously soaked in methanol 100% for 5 min. The blot was blocked with 5% milk (NFDM) in Tris-buffered saline containing 0.1% Tween-20 (TBS-T) for 1 h at room temperature under shaking; next, the membrane was probed overnight at 4 °C with 3D1 mAb as primary antibody at 2 µg/mL in 2.5% NFDM in TBS-T under shaking. The membrane was then triple-washed with TBS-T and incubated with GAM-HRP as secondary antibody diluted 1:1000 in TBS-T for 1 h at room temperature under agitation. The target protein was detected using the enhanced chemi-luminescent substrate method with the SuperSignal West Pico kit provided by Pierce Chemical Co. (Rockford, IL, USA), following the manufacturer’s instructions. Signals were acquired with the ChemiDoc™ XRS video densitometer (Bio-Rad, Hercules, CA, USA), using the Quantity One® software (Bio-Rad, Milano, Italy).
3.14. FACS Analyses
LCP, A375 and WM266 melanoma cells were collected by centrifugation. After aspiration of supernatant, the cells were resuspended in 500 µL of PBS and formaldehyde was added to obtain a final concentration of 4%. Cells were fixed for 10 min at room temperature. After fixation, cells were permeabilized by adding 500 µL of PBS-Tween 20.
1 × 106 cells were aliquoted into each vial; 1 mL of PBS (incubation buffer) was added to each vial and cells were washed by centrifugation at 4000 rpm for 5 min at 4 °C. Cells were resuspended in 100 µL of solution of 3D1 anti-Nodal antibody (primary antibody), diluted in PBS at the following concentrations: 0.1, 0.5, 1.0 and 2.0 µg/mL. A standard IgG1 isotype was used as negative control. Cells were incubated with the primary antibody for 1 h at 4 °C under shaking. After incubation, cells were washed by centrifugation in 1 mL of PBS, resuspended and incubated with a FITC-conjugated anti-mouse antibody (secondary antibody), diluted 1:1000 (1.0 µg/mL) in PBS, for 40 min at 4 °C in the dark under stirring. Cells were washed by centrifugation with 1 mL of incubation buffer, re-suspended in 500 µL of PBS and analyzed on flow cytometer. Fluorescence was evaluated using the BD FACScalibur™ System.
4. Conclusions
Nodal, a member of the TGF-β superfamily, is a potent morphogen and regulator of cell fate in both embryological and adult systems.
Its expression is largely restricted to very early progenitor and reproductive cell types, whereas it is almost absent in adult tissues and normal cells, including melanocytes. Its re-expression in the adults is strongly associated with tumorigenesis, indeed Nodal levels are much higher in tumor cells and tissues where it supports uncontrolled cell growth, proliferation and differentiation [
8].
Several studies have reported that Nodal expression positively correlates with melanoma tumor progression toward a metastatic phenotype; moreover, its over-expression is paralleled by that of the co-receptor Cripto-1, suggesting the occurrence of tumor growth mechanisms supported by the concomitant presence of both proteins and by their mutual interaction with the activin-like receptor complex [
5,
9,
10,
11,
12,
13,
22,
23].
In light of this evidence, Nodal is considered a promising diagnostic and prognostic marker and a very important and interesting new therapeutic target for several types of cancers [
4]. So far, exogenous Nodal inhibitors contrasting its anti-apoptotic and tumorigenic activity have not been reported. Nodal activity down-regulation, which dampens both Cripto-1-dependent and independent signalling, has been only achieved by using antisense technology, an approach that has largely contributed to elucidate melanoma-related functions [
9].
To develop pharmaceutical inhibitors and reagents useful for its detection in tumor tissues, we have targeted Nodal with monoclonal antibodies
ad hoc generated to bind the Cripto-1 interaction surface. mAbs have been developed against a Nodal peptide encompassing residues 44–67 [
21,
31].
We have privileged the approach based on mAbs because they are readily accessible by the hybridoma technology, which generally affords antibodies in a straightforward manner and in high amounts. Antibodies from supernatants of stabilized hybridoma cells are also easily purified and characterized by current chromatographic techniques and, most importantly, are in most cases endowed with a sufficiently high affinity and specificity for the target antigens.
The anti-Nodal mAbs have been generated and screened following a novel, subtractive approach to select only those able to bind the wild type peptide antigen and not a variant mutated containing two mutated alanine on E49 and E50, two specific residues directly involved in the binding with Cripto-1 [
21,
31]. mAbs selected by this approach had the potential to interact with high specificity with these and other surrounding residues thereby working as both Nodal binders and inhibitors of the interaction with Cripto-1. Antibody 3D1 also recognized with high affinity the full-length native protein. The ability of 3D1 to recognize native and non-native Nodal under different experimental conditions has been here proven by a variety of techniques, therefore the antibody can be seen as a new analytical tool for detecting the protein in tissues and in biological fluids where it is expressed and in theragnostic programs aimed at developing Nodal inhibitors. Remarkably, 3D1 also blocks
in vitro the interaction of Nodal with Cripto-1, a feature of particular relevance in the context of tumor development mechanisms involving the activation of the Smad2/3 axis [
32]. As recently shown [
20,
21], Nodal also binds to ALK4 and the region of binding seems to be shared with Cripto-1 and ALK7, two well-established Nodal (co)-receptors. On this basis we could speculate that 3D1 has the potential to also inhibit the interaction of Nodal with other receptors thus strengthening inhibition of Smad 2/3 activation and potentiating the anti-tumoral activity of the antibody [
32].
Collectively analytical and functional data show that the anti-Nodal 3D1 mAb is a promising reagent for the detection of Nodal under different conditions and in different tissues. The Smad 2/3 and MAPK neutralizing activity also suggests it is a promising novel biotherapeutic agent.
The generation of smaller functional fragments (Fab, ScFv), together with a step of affinity improvement and humanization, will provide a unique and invaluable reagent for the treatment, alone or in combination with other drugs, of cutaneous melanoma and other relevant cancer diseases.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




