
Adaptive Immune Responses in a Multiple Sclerosis Patient with Acute Varicella-Zoster Virus Reactivation during Treatment with Fingolimod

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
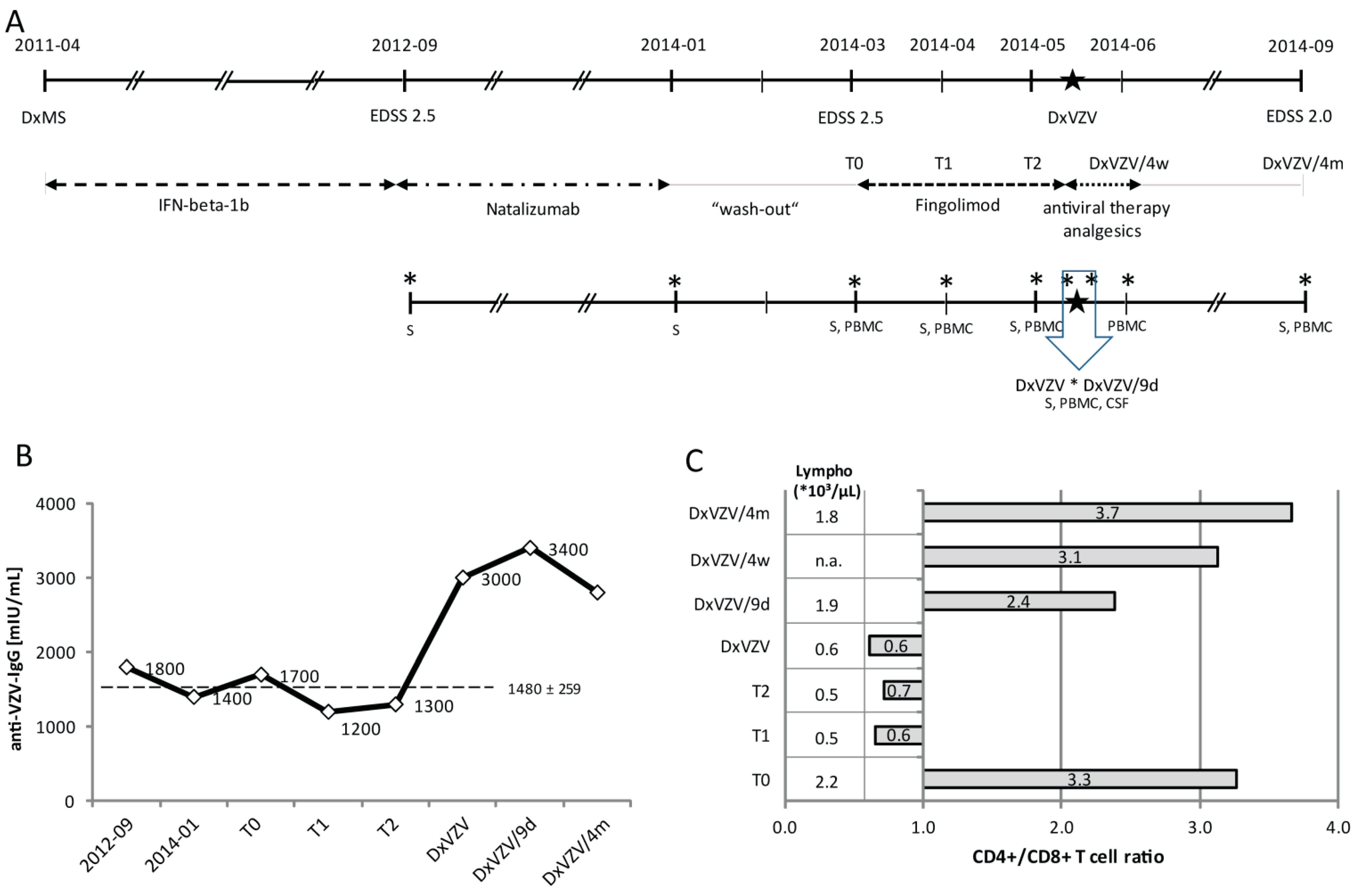
2.1.1. Clinical Case Report

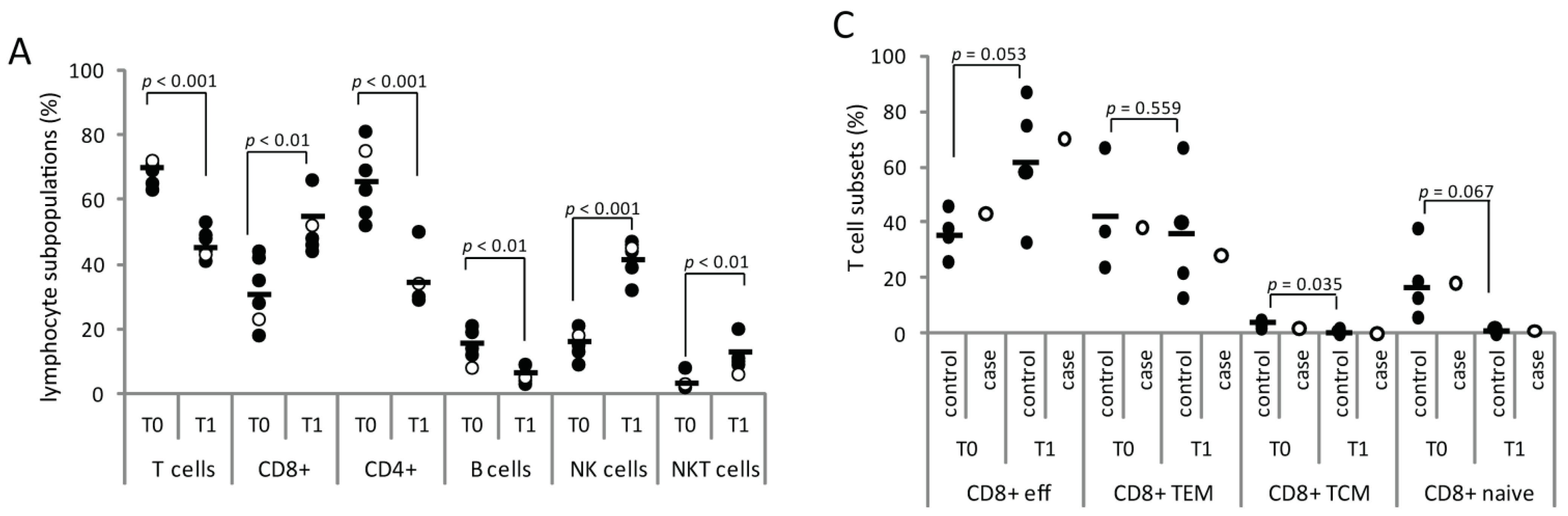
2.1.2. Serial Analysis of Humoral and Cellular Immune Parameters


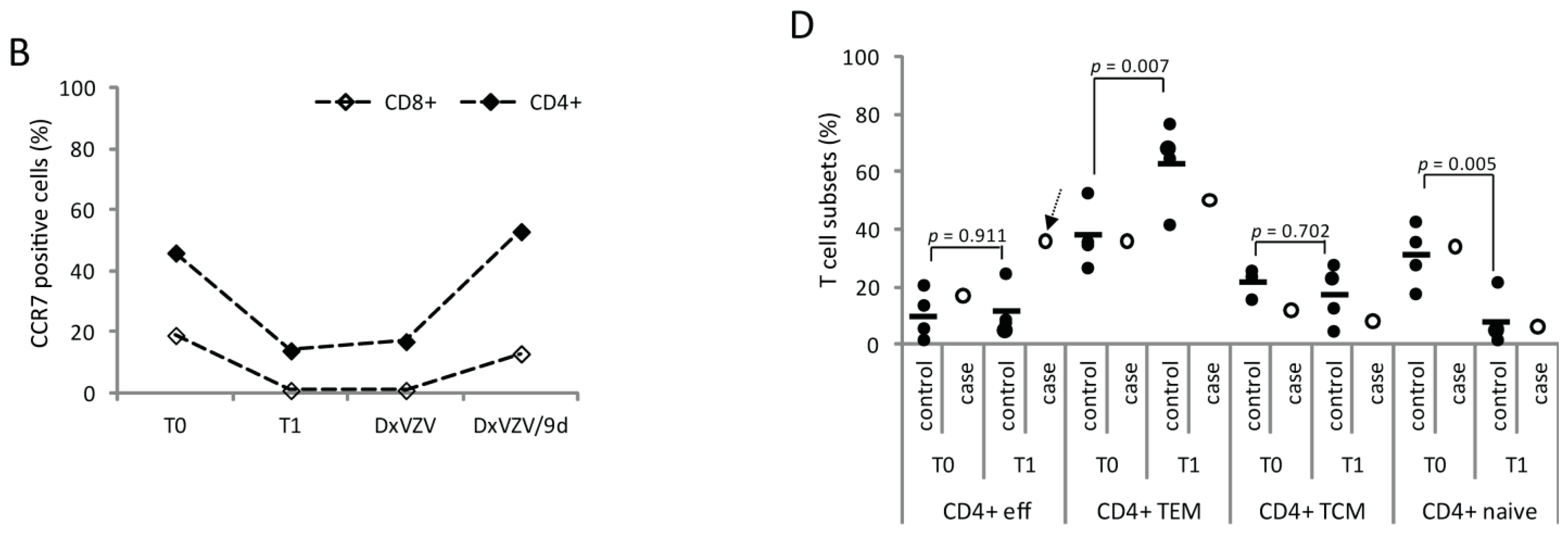
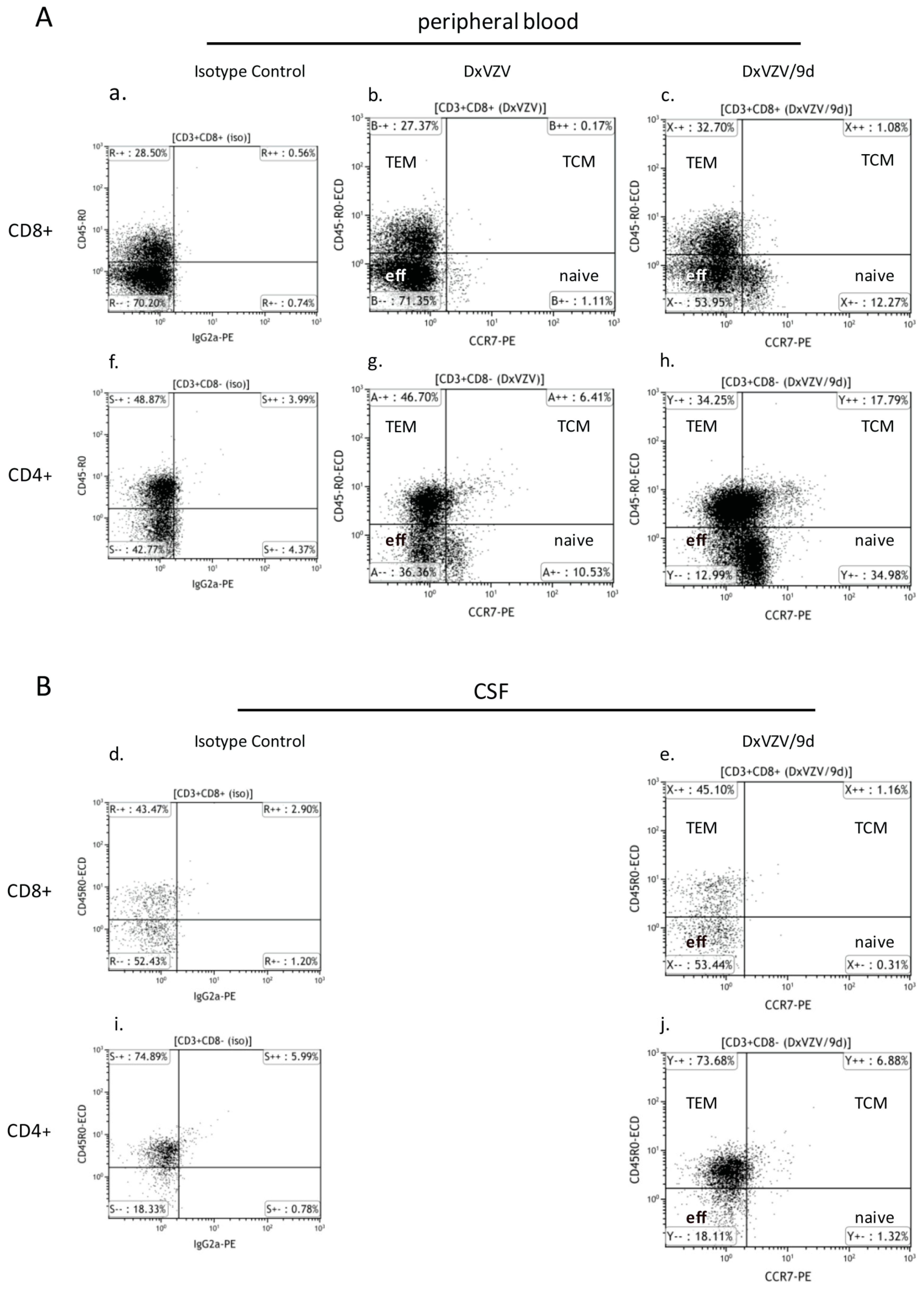
2.1.3. Cellular Immune Parameters and Immune Phenotypes of Peripheral Blood and Cerebrospinal Fluid (CSF) Cells during Intravenous Acyclovir and Fingolimod (FTY) Discontinuation

2.2. Discussion
3. Experimental Section
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bader, M.S. Herpes zoster: Diagnostic, therapeutic, and preventive approaches. Postgrad. Med. 2013, 125, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Nagel, M.A.; Gilden, D. The challenging patient with varicella-zoster virus disease. Neurol. Clin. Pract. 2013, 3, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, A.; Levin, M.J. VZV T cell-mediated immunity. Curr. Top. Microbiol. Immunol. 2010, 342, 341–357. [Google Scholar] [PubMed]
- Brinkmann, V. FTY720 (fingolimod) in Multiple Sclerosis: Therapeutic effects in the immune and the central nervous system. Br. J. Pharmacol. 2009, 158, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Mehling, M.; Brinkmann, V.; Antel, J.; Bar-Or, A.; Goebels, N.; Vedrine, C.; Kristofic, C.; Kuhle, J.; Lindberg, R.L.; Kappos, L. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology 2008, 71, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Radue, E.W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.M.; Baumgartner, A.; Rauer, S.; Stich, O. Multiple sclerosis rebound following herpes zoster infection and suspension of fingolimod. Neurology 2012, 79, 2006–2007. [Google Scholar] [CrossRef] [PubMed]
- Issa, N.P.; Hentati, A. VZV encephalitis that developed in an immunized patient during fingolimod therapy. Neurology 2015, 84, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Ratchford, J.N.; Costello, K.; Reich, D.S.; Calabresi, P.A. Varicella-zoster virus encephalitis and vasculopathy in a patient treated with fingolimod. Neurology 2013, 81, 306. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Wolinsky, J.S.; Kappos, L.; Morris, M.I.; Reder, A.T.; Tornatore, C.; Gershon, A.; Gershon, M.; Levin, M.J.; Bezuidenhoudt, M.; et al. Varicella-zoster virus infections in patients treated with fingolimod: Risk assessment and consensus recommendations for management. JAMA Neurol. 2015, 72, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, M.E.; Lorscheider, J.; Waschbisch, A.; Paroz, C.; Mehta, S.K.; Pierson, D.L.; Kuhle, J.; Fischer-Barnicol, B.; Sprenger, T.; Lindberg, R.L.; et al. T-cell response against varicella-zoster virus in fingolimod-treated MS patients. Neurology 2013, 81, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, H.; Sharp, M.; Maecker, H.T.; Maino, V.C.; Arvin, A.M. Frequencies of memory T cells specific for varicella-zoster virus, herpes simplex virus, and cytomegalovirus by intracellular detection of cytokine expression. J. Infect. Dis. 2000, 181, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L. Fingolimod and risk of varicella-zoster virus infection: Back to the future with an old infection and a new drug. JAMA Neurol. 2015, 72, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Hufner, K.; Derfuss, T.; Herberger, S.; Sunami, K.; Russell, S.; Sinicina, I.; Arbusow, V.; Strupp, M.; Brandt, T.; Theil, D. Latency of alpha-herpes viruses is accompanied by a chronic inflammation in human trigeminal ganglia but not in dorsal root ganglia. J. Neuropathol. Exp. Neurol. 2006, 65, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Champagne, P.; Ogg, G.S.; King, A.S.; Knabenhans, C.; Ellefsen, K.; Nobile, M.; Appay, V.; Rizzardi, G.P.; Fleury, S.; Lipp, M.; et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature 2001, 410, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Ntranos, A.; Hall, O.; Robinson, D.P.; Grishkan, I.V.; Schott, J.T.; Tosi, D.M.; Klein, S.L.; Calabresi, P.A.; Gocke, A.R. FTY720 impairs CD8 T-cell function independently of the sphingosine-1-phosphate pathway. J. Neuroimmunol. 2014, 270, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Schub, D.; Janssen, E.; Leyking, S.; Sester, U.; Assmann, G.; Hennes, P.; Smola, S.; Vogt, T.; Rohrer, T.; Sester, M.; et al. Altered phenotype and functionality of varicella zoster virus-specific cellular immunity in individuals with active infection. J. Infect. Dis. 2015, 211, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Malavige, G.N.; Jones, L.; Black, A.P.; Ogg, G.S. Varicella zoster virus glycoprotein E-specific CD4+ T cells show evidence of recent activation and effector differentiation, consistent with frequent exposure to replicative cycle antigens in healthy immune donors. Clin. Exp. Immunol. 2008, 152, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Boulton, C.; Meiser, K.; David, O.J.; Schmouder, R. Pharmacodynamic effects of steady-state fingolimod on antibody response in healthy volunteers: A 4-week, randomized, placebo-controlled, parallel-group, multiple-dose study. J. Clin. Pharmacol. 2012, 52, 1879–1890. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Mehling, M.; Arroyo, R.; Izquierdo, G.; Selmaj, K.; Curovic-Perisic, V.; Keil, A.; Bijarnia, M.; Singh, A.; von Rosenstiel, P. Randomized trial of vaccination in fingolimod-treated patients with multiple sclerosis. Neurology 2015, 84, 872–879. [Google Scholar] [CrossRef] [PubMed]
- David, O.J.; Kovarik, J.M.; Schmouder, R.L. Clinical pharmacokinetics of fingolimod. Clin. Pharmacokinet. 2012, 51, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zollinger, M.; Borell, H.; Zimmerlin, A.; Patten, C.J. CYP4F enzymes are responsible for the elimination of fingolimod (FTY720), a novel treatment of relapsing multiple sclerosis. Drug Metab. Dispos. 2011, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Zollinger, M.; Gschwind, H.P.; Jin, Y.; Sayer, C.; Zecri, F.; Hartmann, S. Absorption and disposition of the sphingosine 1-phosphate receptor modulator fingolimod (FTY720) in healthy volunteers: A case of xenobiotic biotransformation following endogenous metabolic pathways. Drug Metab. Dispos. 2011, 39, 199–207. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Available online: http://www.fda.gov/Drugs (accessed on 15 July 2015).
- Mueller, S.N.; Hosiawa-Meagher, K.A.; Konieczny, B.T.; Sullivan, B.M.; Bachmann, M.F.; Locksley, R.M.; Ahmed, R.; Matloubian, M. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science 2007, 317, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Pellkofer, H.L.; Cepok, S.; Korn, T.; Kumpfel, T.; Buck, D.; Hohlfeld, R.; Berthele, A.; Hemmer, B. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology 2011, 76, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Roesler, J.; Hedrich, C.; Laass, M.W.; Heyne, K.; Rosen-Wolff, A. Meningoencephalitis caused by varicella-zoster virus reactivation in a child with dominant partial interferon-γ receptor-1 deficiency. Pediatr. Infect. Dis. J. 2011, 30, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, Y.; Cavanagh, M.M.; Le, S.S.; Qi, Q.; Roskin, K.M.; Looney, T.J.; Lee, J.Y.; Dixit, V.; Dekker, C.L.; et al. B-cell repertoire responses to varicella-zoster vaccination in human identical twins. Proc. Natl. Acad. Sci. USA 2015, 112, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Arvin, A. Investigation of varicella-zoster virus neurotropism and neurovirulence using SCID mouse-human DRG xenografts. J. Neurovirol. 2011, 17, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Serpero, L.D.; Filaci, G.; Parodi, A.; Battaglia, F.; Kalli, F.; Brogi, D.; Mancardi, G.L.; Uccelli, A.; Fenoglio, D. Fingolimod modulates peripheral effector and regulatory T cells in MS patients. J. Neuroimmune. Pharmacol. 2013, 8, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Mehling, M.; Hilbert, P.; Fritz, S.; Durovic, B.; Eichin, D.; Gasser, O.; Kuhle, J.; Klimkait, T.; Lindberg, R.L.; Kappos, L.; et al. Antigen-specific adaptive immune responses in fingolimod-treated multiple sclerosis patients. Ann. Neurol. 2011, 69, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, E.; Dubois, G.; Jarai, G.; Edwards, M.; Thomas, M.; Nicholls, A.; Albert, R.; Newson, C.; Brinkmann, V.; Walker, C. The sphingosine 1-phosphate receptor agonist FTY720 differentially affects the sequestration of CD4+/CD25+ T-regulatory cells and enhances their functional activity. J. Immunol. 2005, 175, 7973–7980. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, D.; de Biasi, S.; Vitetta, F.; Simone, A.M.; Federzoni, L.; Borghi, V.; Cossarizza, A.; Nichelli, P.F.; Sola, P. Recurrent varicella following steroids and fingolimod in a multiple sclerosis patient. J. Neuroimmune. Pharmacol. 2013, 8, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Debiasi, R.L.; Tyler, K.L. Molecular methods for diagnosis of viral encephalitis. Clin. Microbiol. Rev. 2004, 17, 903–925. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrer, A.; Wipfler, P.; Pilz, G.; Oppermann, K.; Haschke-Becher, E.; Afazel, S.; Kraus, J.; Trinka, E.; Sellner, J. Adaptive Immune Responses in a Multiple Sclerosis Patient with Acute Varicella-Zoster Virus Reactivation during Treatment with Fingolimod. Int. J. Mol. Sci. 2015, 16, 21832-21845. https://doi.org/10.3390/ijms160921832
Harrer A, Wipfler P, Pilz G, Oppermann K, Haschke-Becher E, Afazel S, Kraus J, Trinka E, Sellner J. Adaptive Immune Responses in a Multiple Sclerosis Patient with Acute Varicella-Zoster Virus Reactivation during Treatment with Fingolimod. International Journal of Molecular Sciences. 2015; 16(9):21832-21845. https://doi.org/10.3390/ijms160921832
Chicago/Turabian StyleHarrer, Andrea, Peter Wipfler, Georg Pilz, Katrin Oppermann, Elisabeth Haschke-Becher, Shahrzad Afazel, Jörg Kraus, Eugen Trinka, and Johann Sellner. 2015. "Adaptive Immune Responses in a Multiple Sclerosis Patient with Acute Varicella-Zoster Virus Reactivation during Treatment with Fingolimod" International Journal of Molecular Sciences 16, no. 9: 21832-21845. https://doi.org/10.3390/ijms160921832
APA StyleHarrer, A., Wipfler, P., Pilz, G., Oppermann, K., Haschke-Becher, E., Afazel, S., Kraus, J., Trinka, E., & Sellner, J. (2015). Adaptive Immune Responses in a Multiple Sclerosis Patient with Acute Varicella-Zoster Virus Reactivation during Treatment with Fingolimod. International Journal of Molecular Sciences, 16(9), 21832-21845. https://doi.org/10.3390/ijms160921832