
Modulation of the PI3K/Akt Pathway and Bcl-2 Family Proteins Involved in Chicken’s Tubular Apoptosis Induced by Nickel Chloride (NiCl2)

 ,
,
Abstract
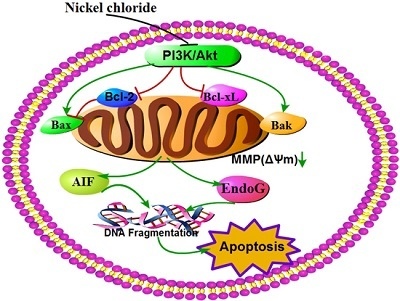
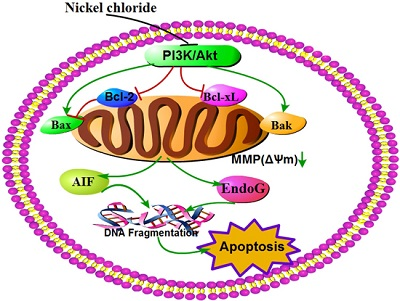
:
1. Introduction
2. Results
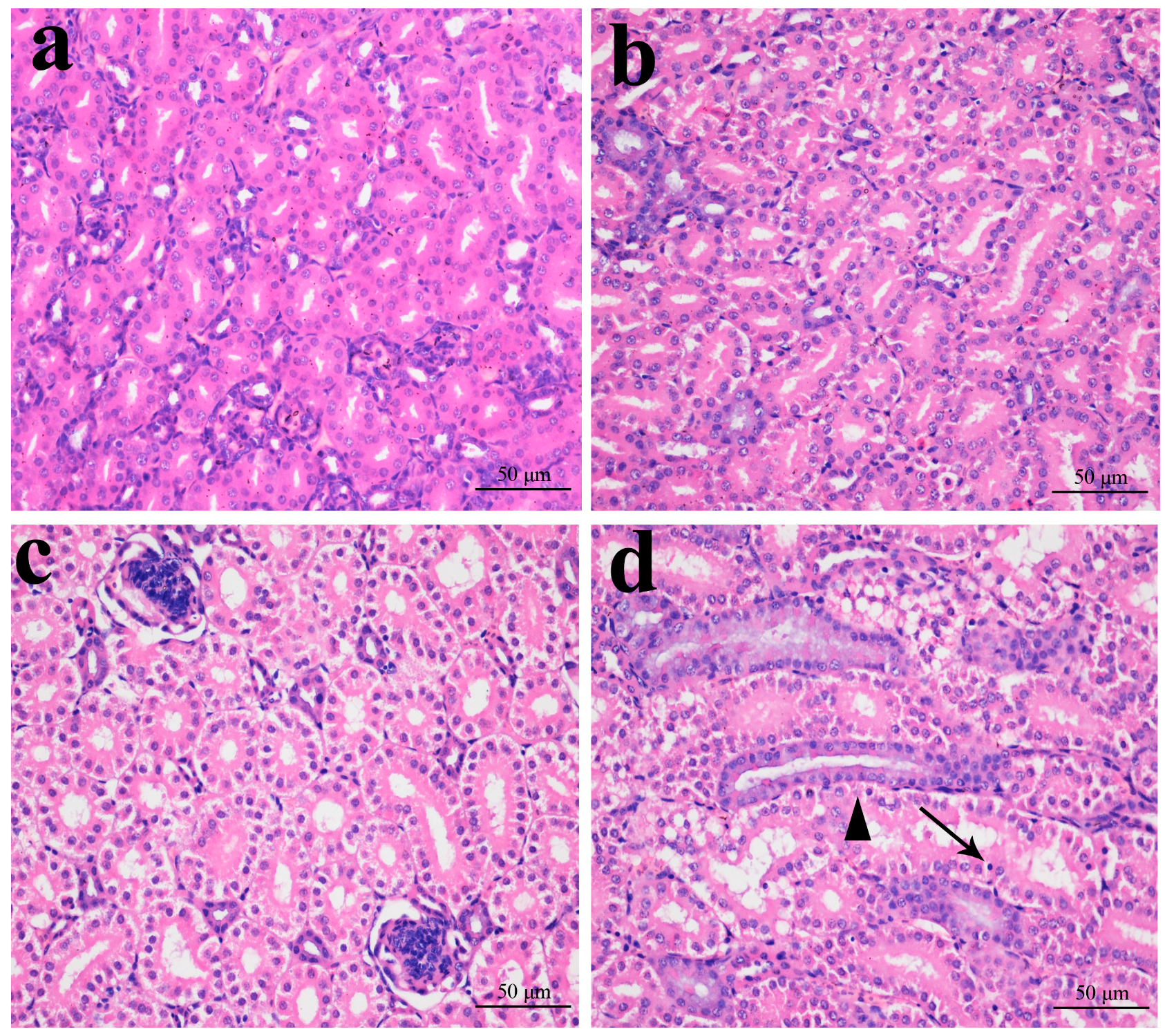
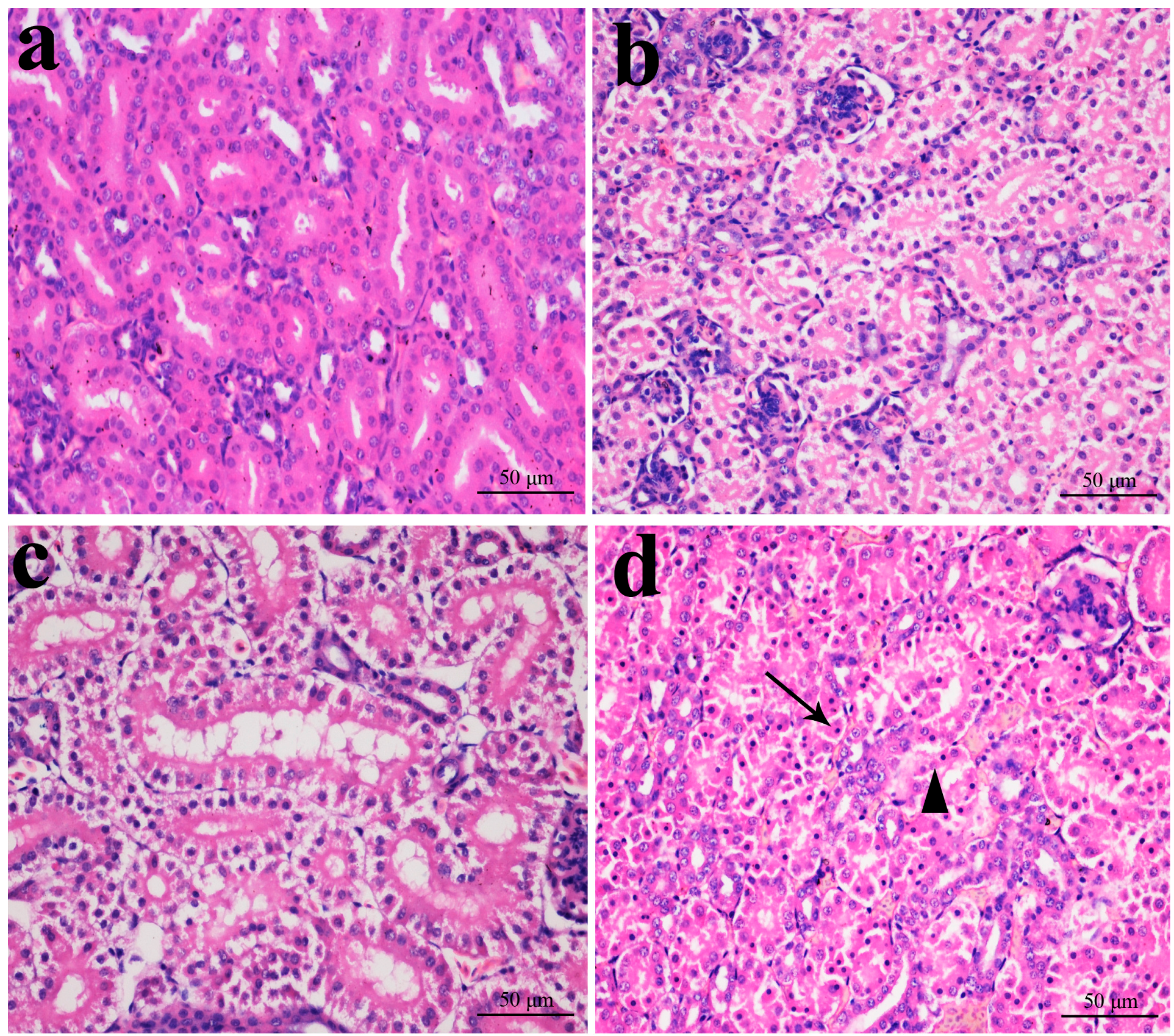
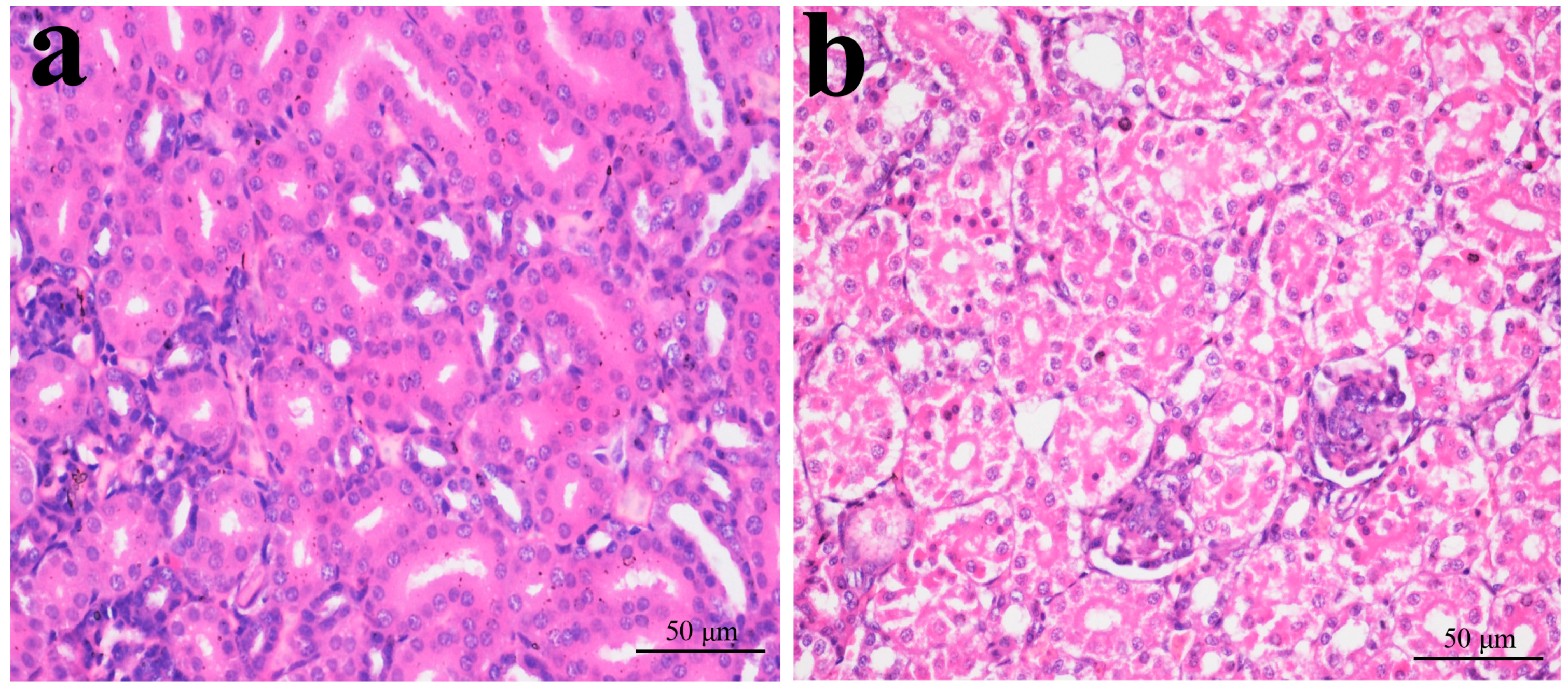
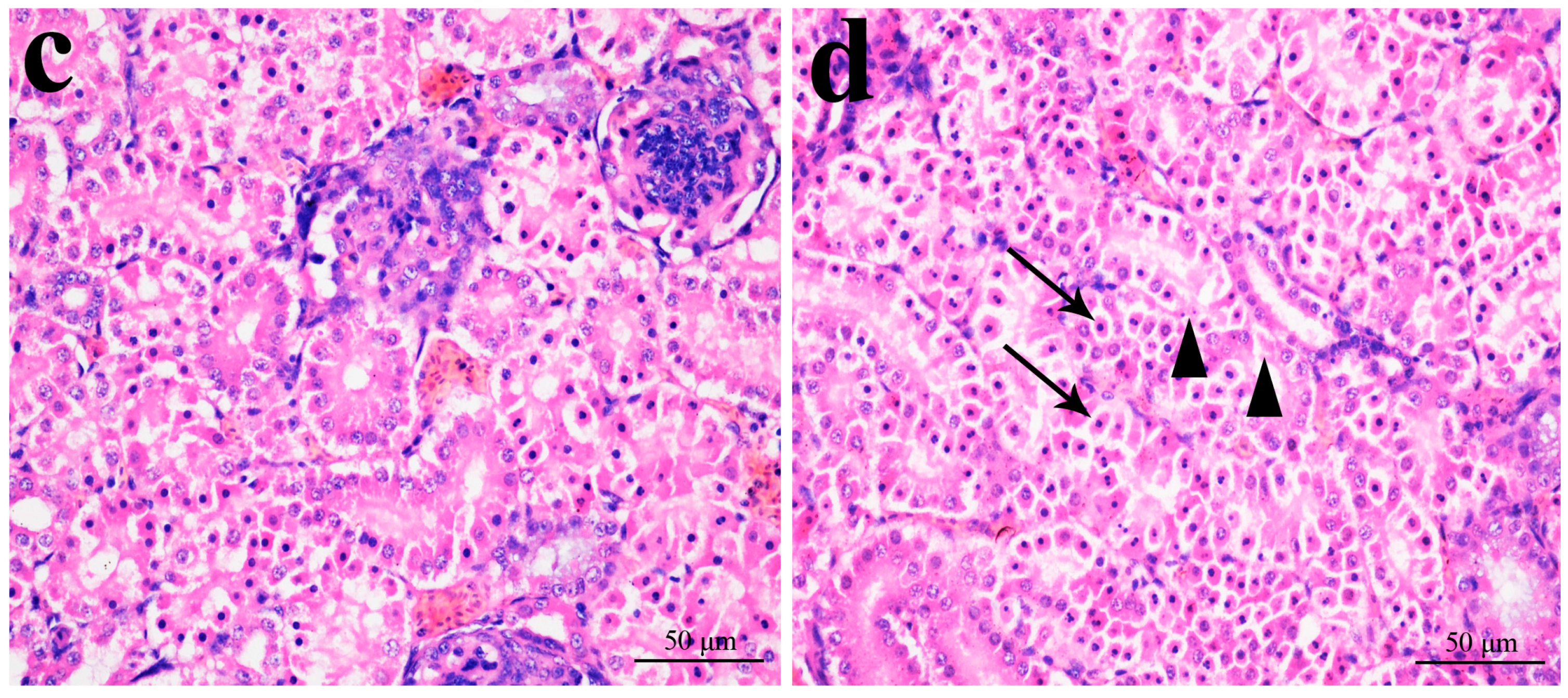
2.1. Histopathological Changes in the Kidney





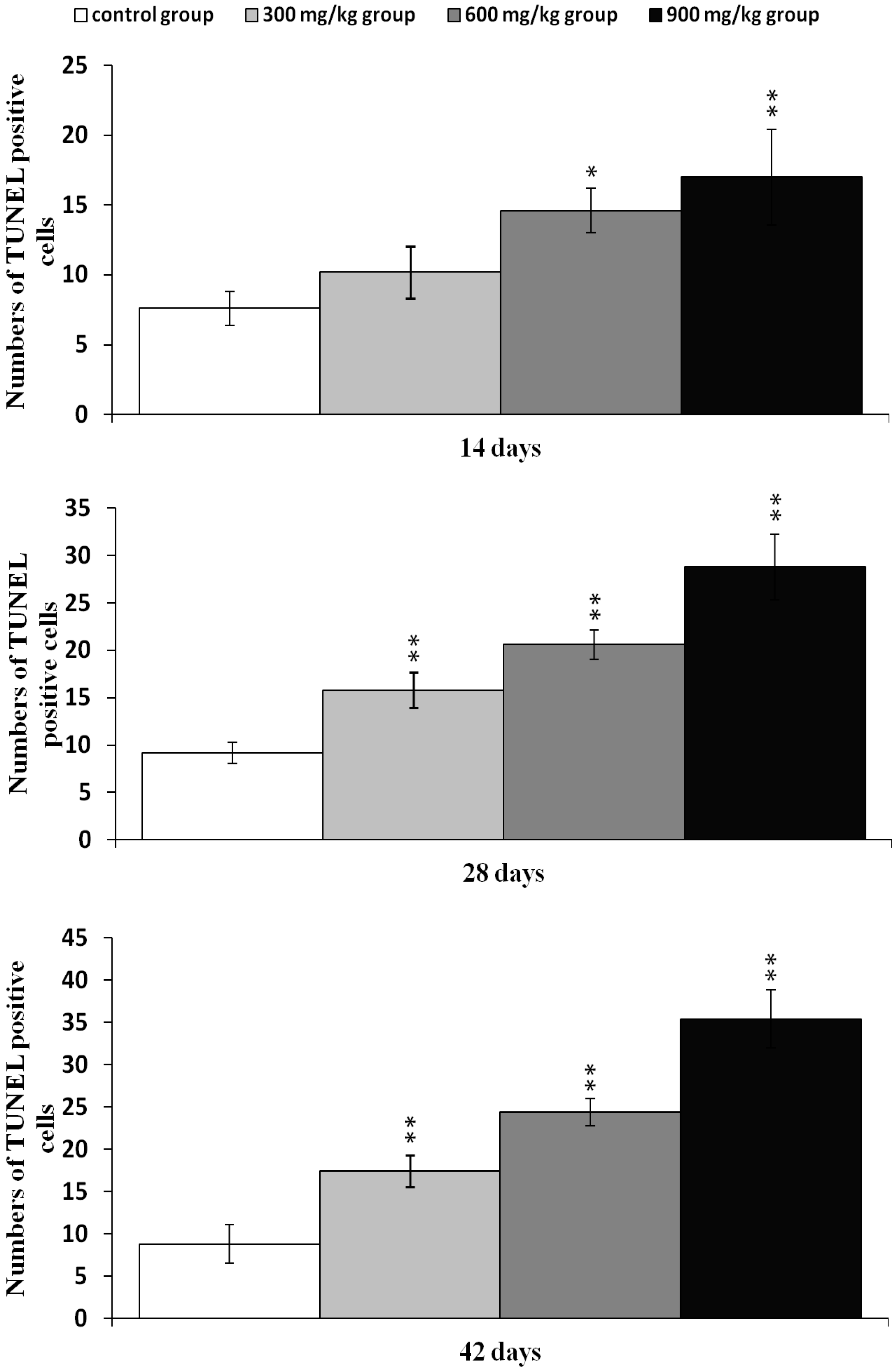
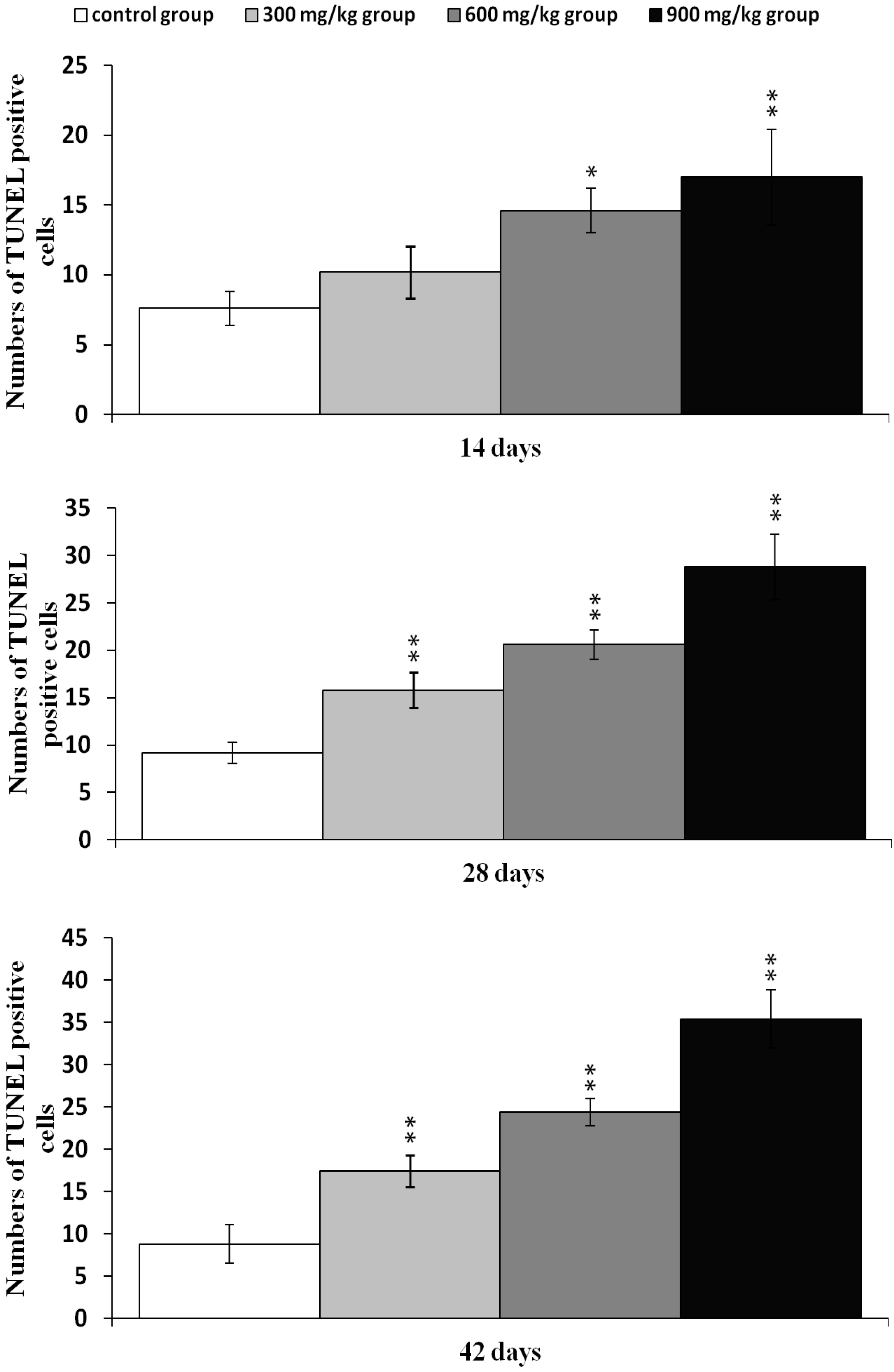
2.2. Effects of NiCl2 on Apoptosis in the Kidney
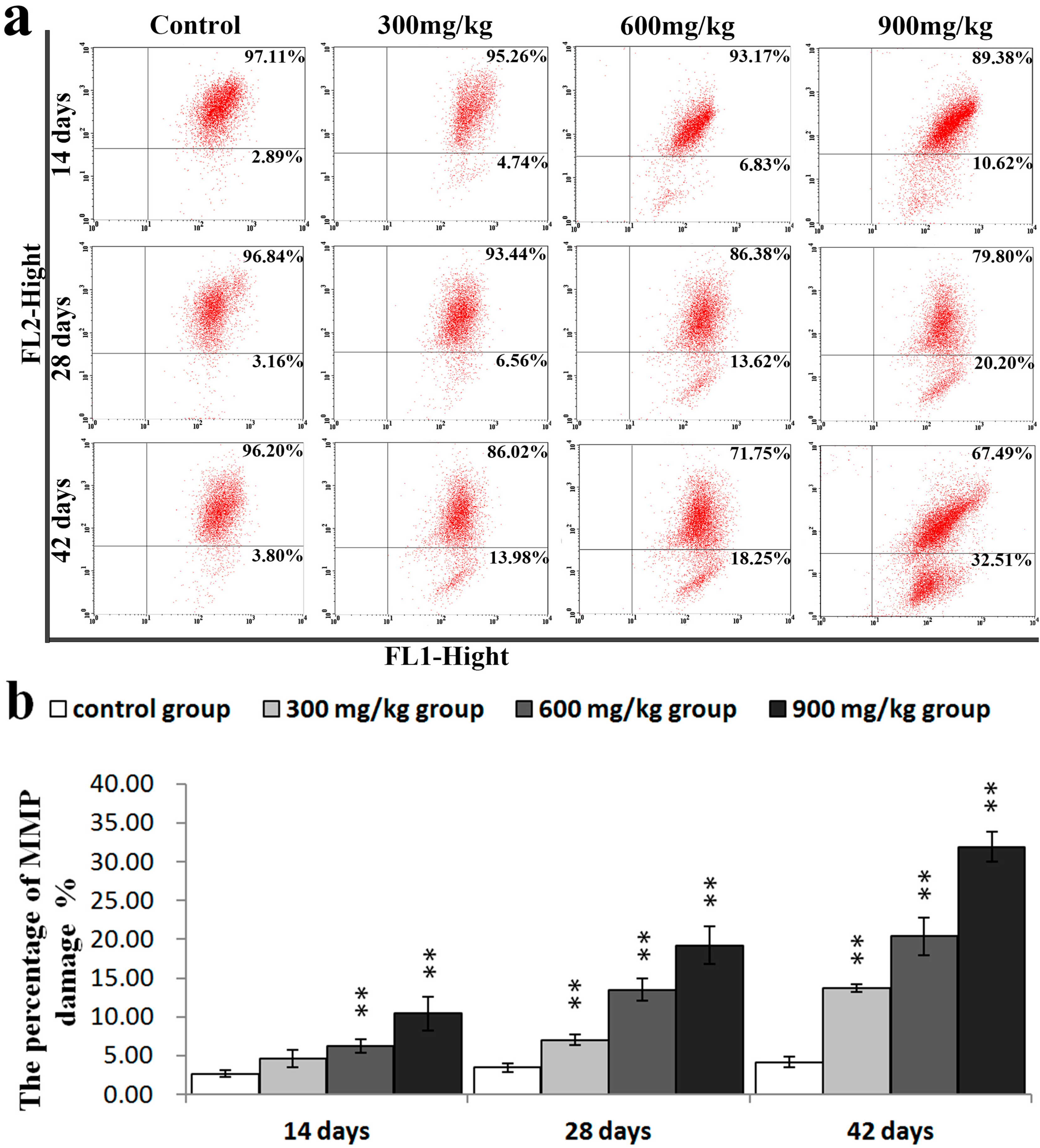
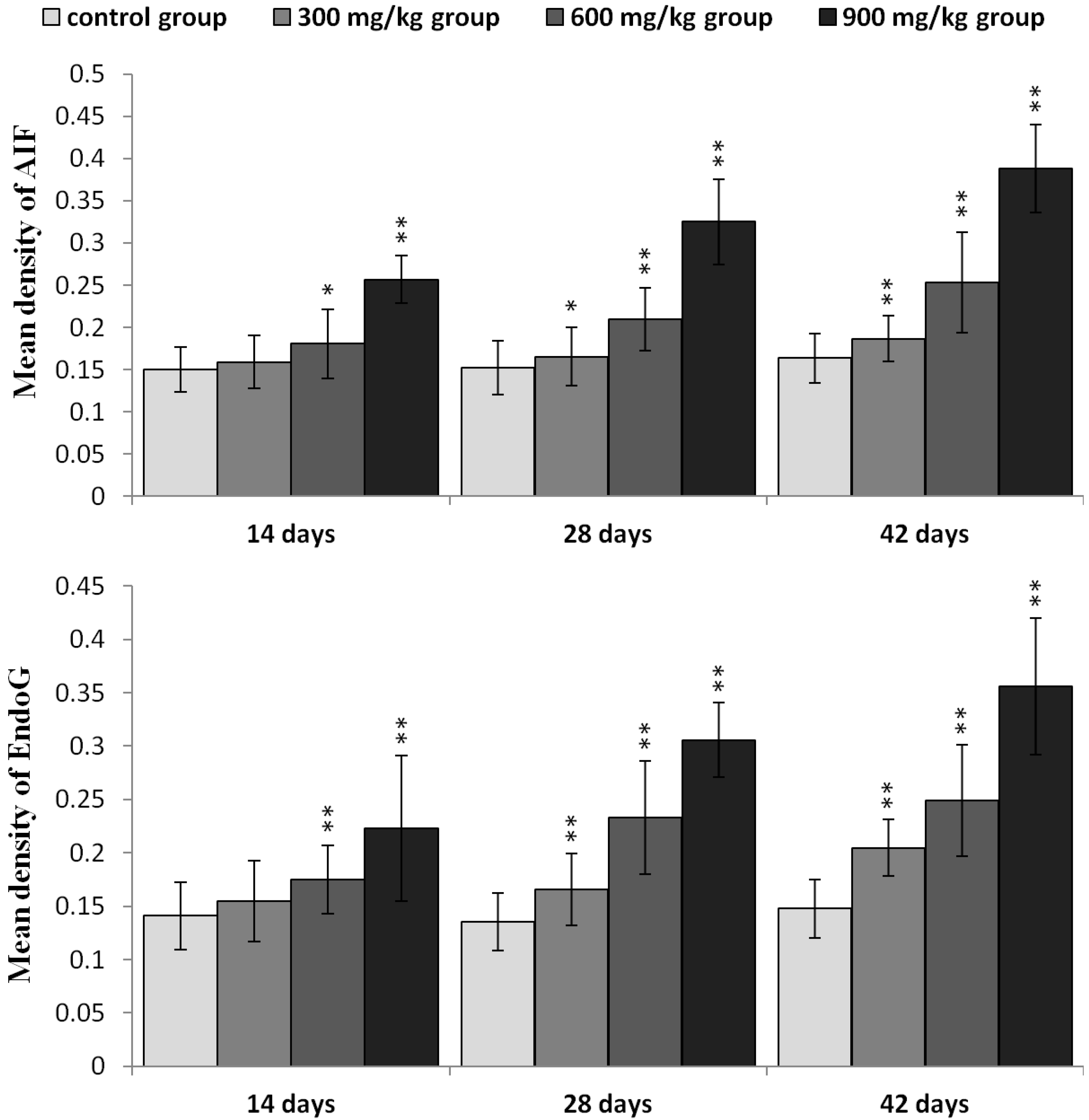
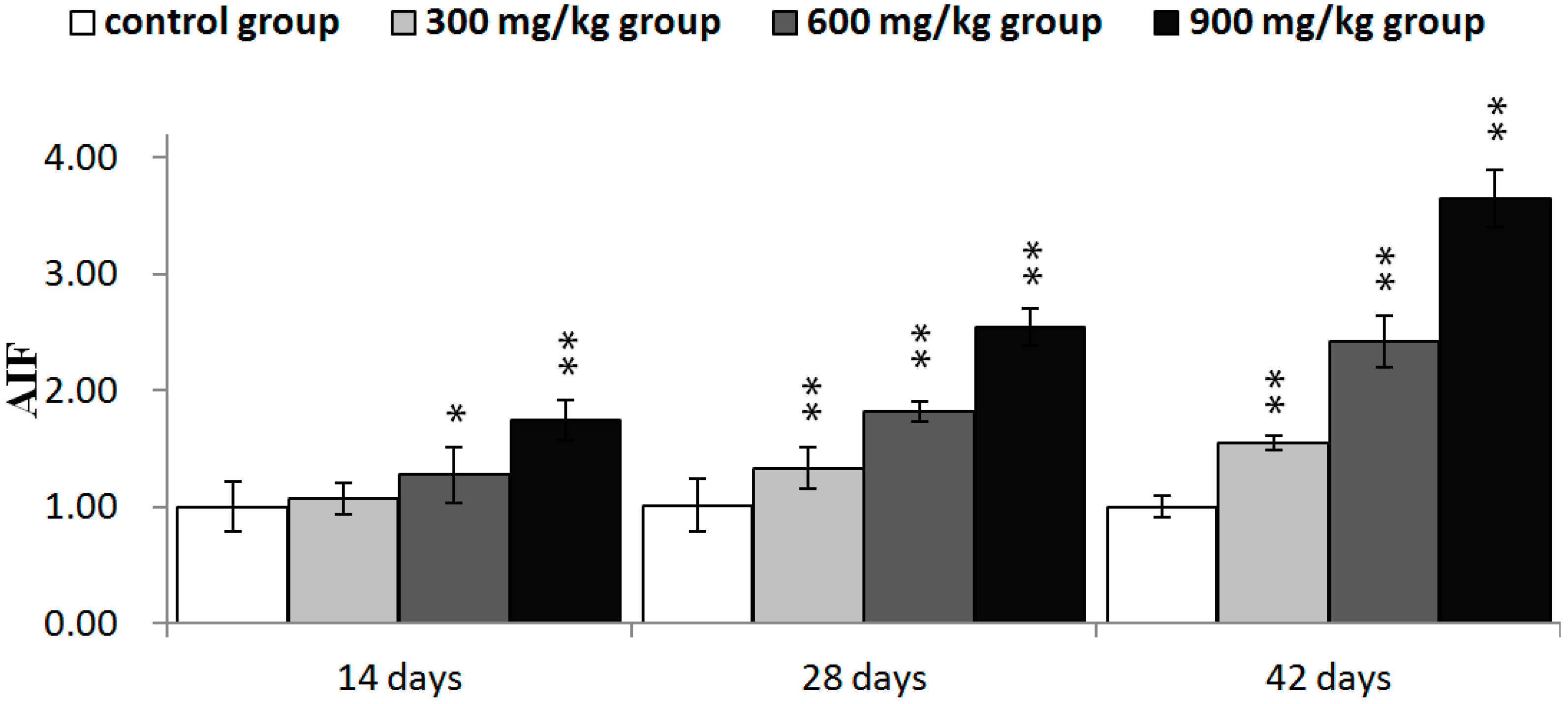
2.3. Effects of NiCl2 on MMP, and AIF and EndoG Protein and mRNA Expression in the Kidney




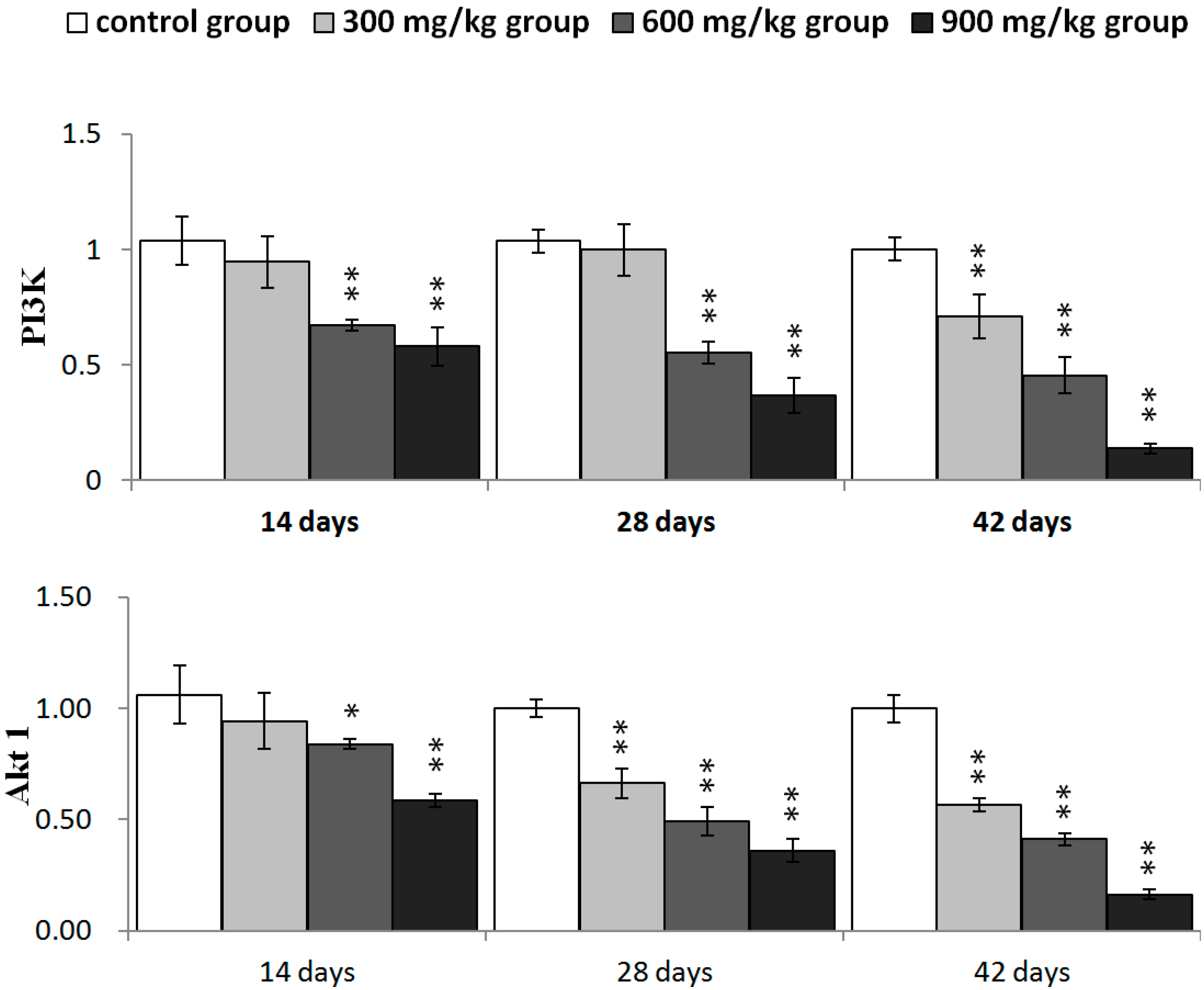
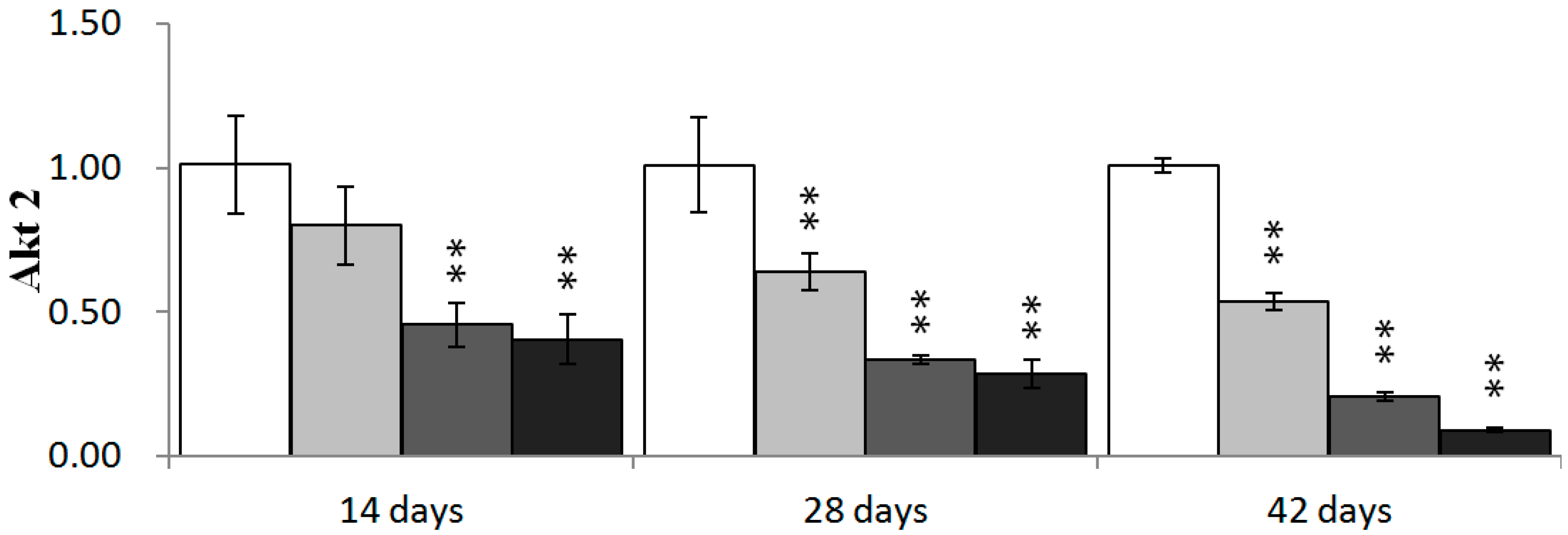
2.4. Effects of NiCl2 on Phosphoinositide-3-Kinase (PI3K)/Serine-Threonine Kinase (Akt) Pathway in the Kidney


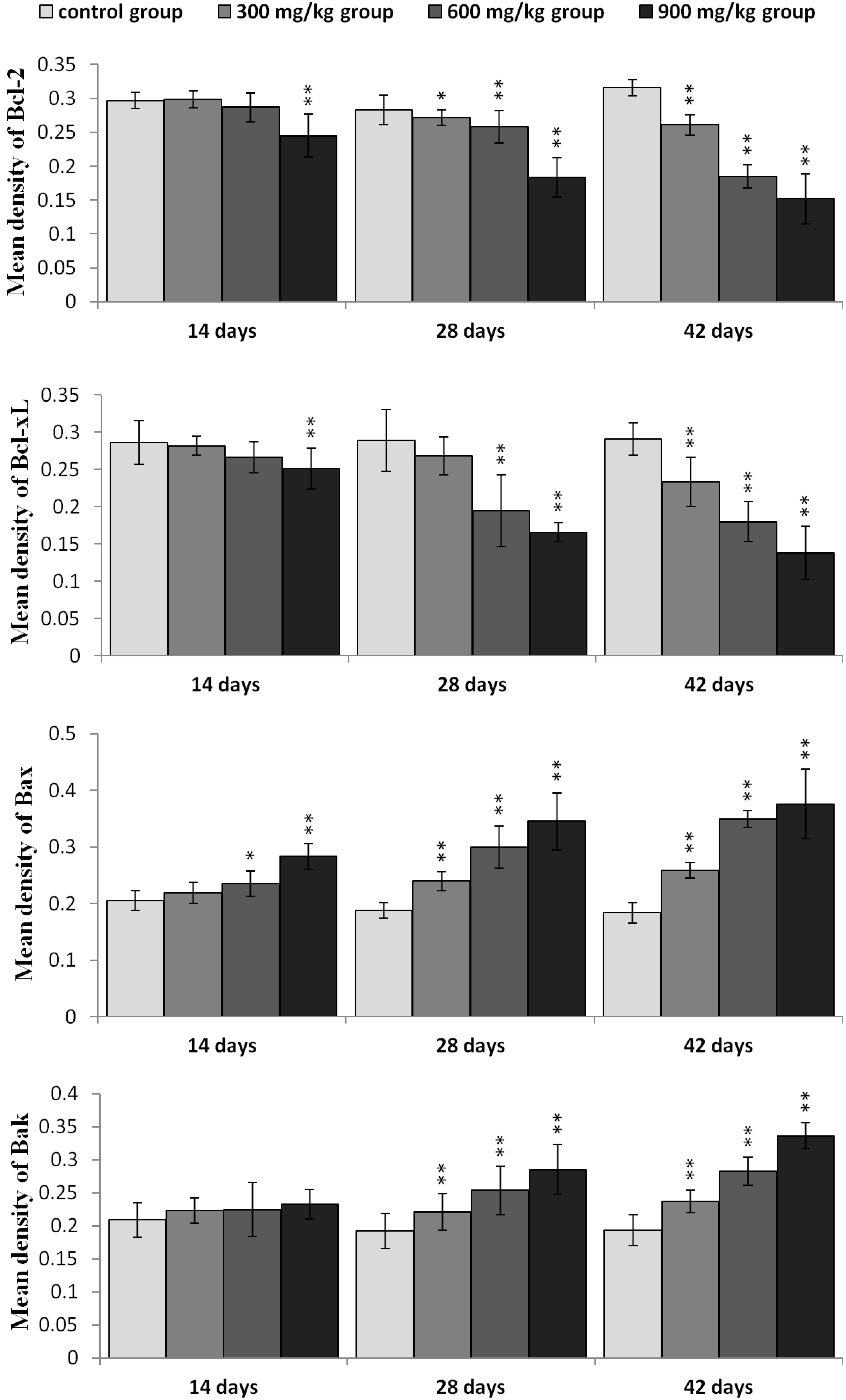
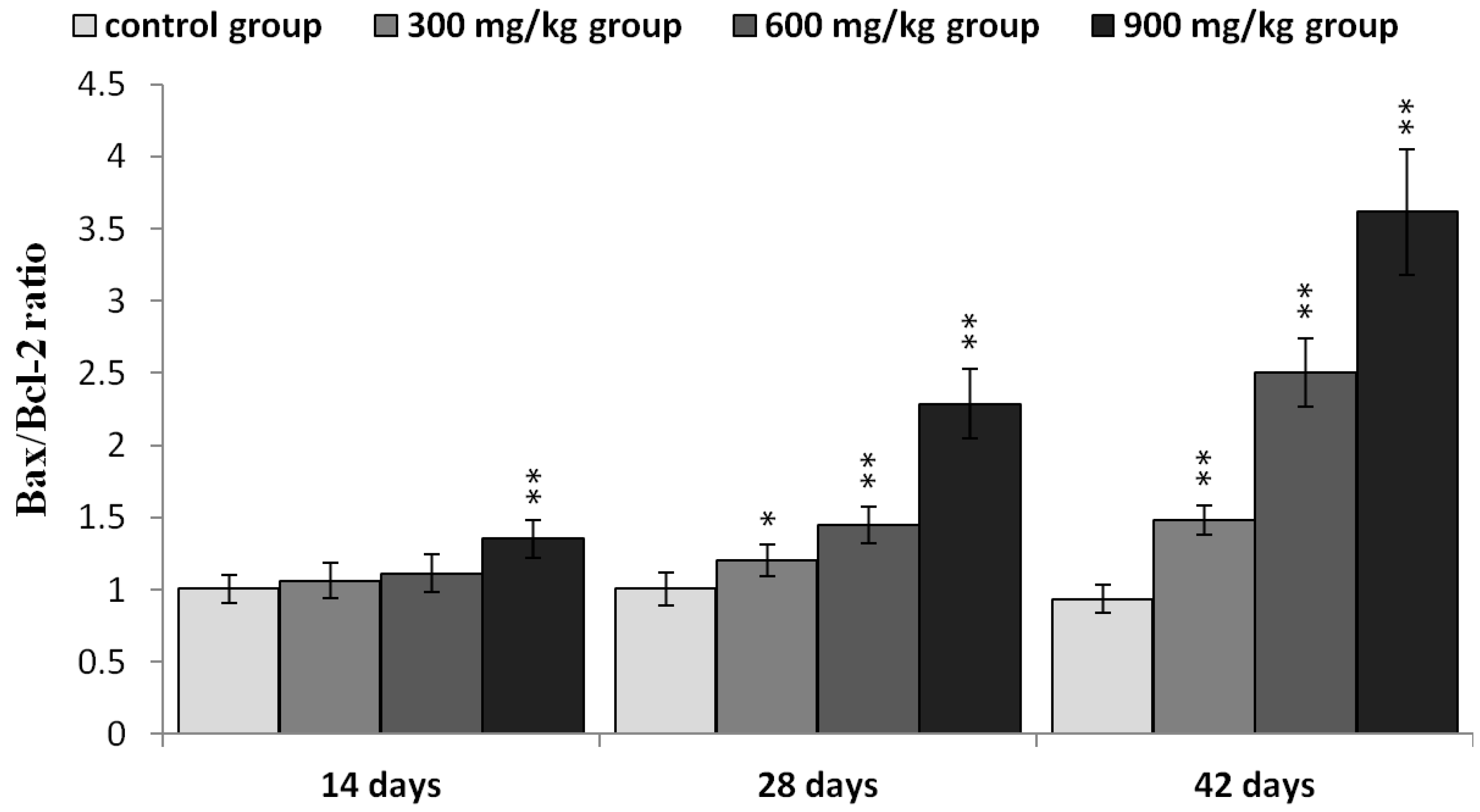
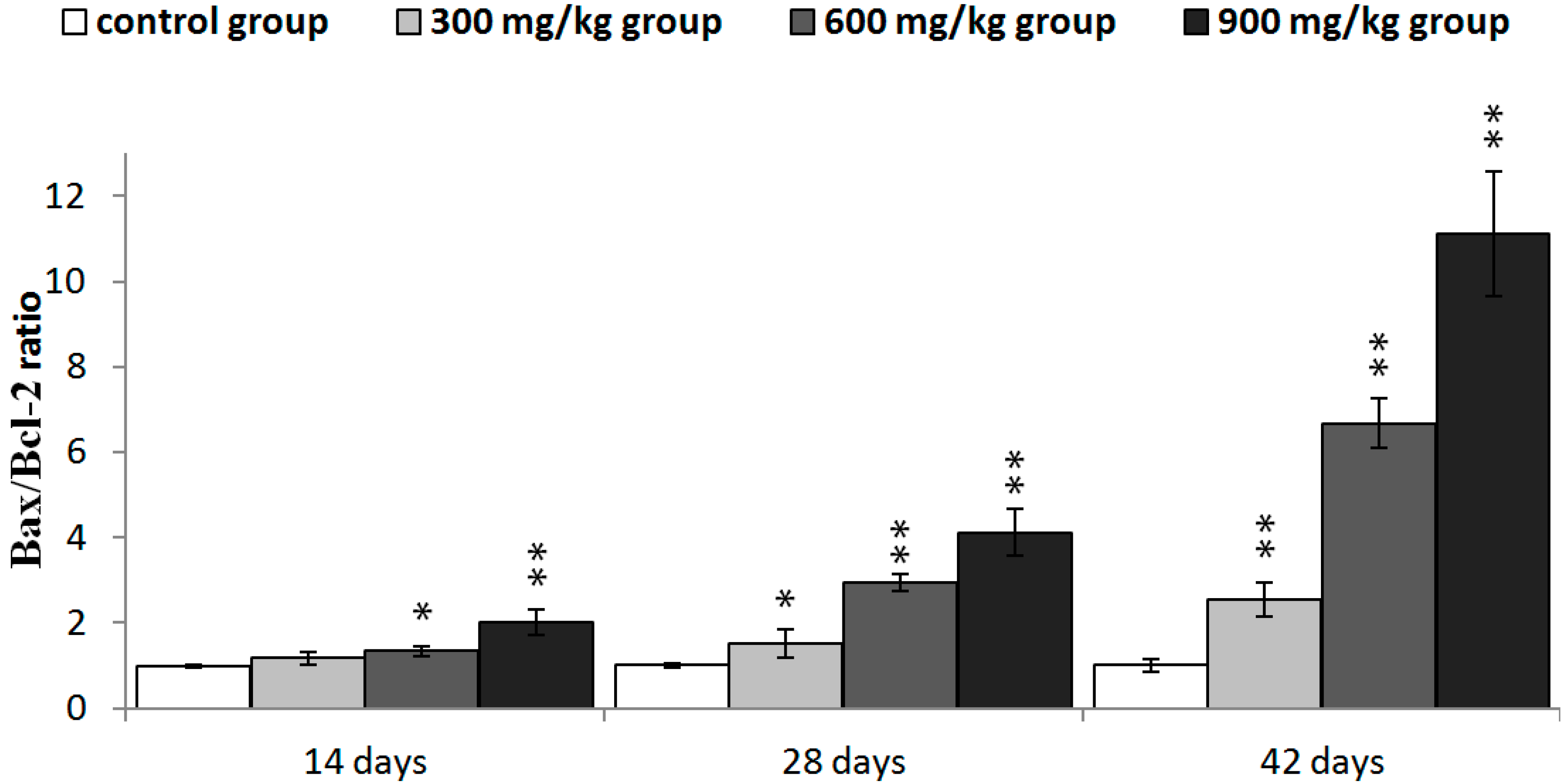
2.5. Expression of NiCl2 on Bcl-2 Family Protein and mRNA Expression in the Kidney





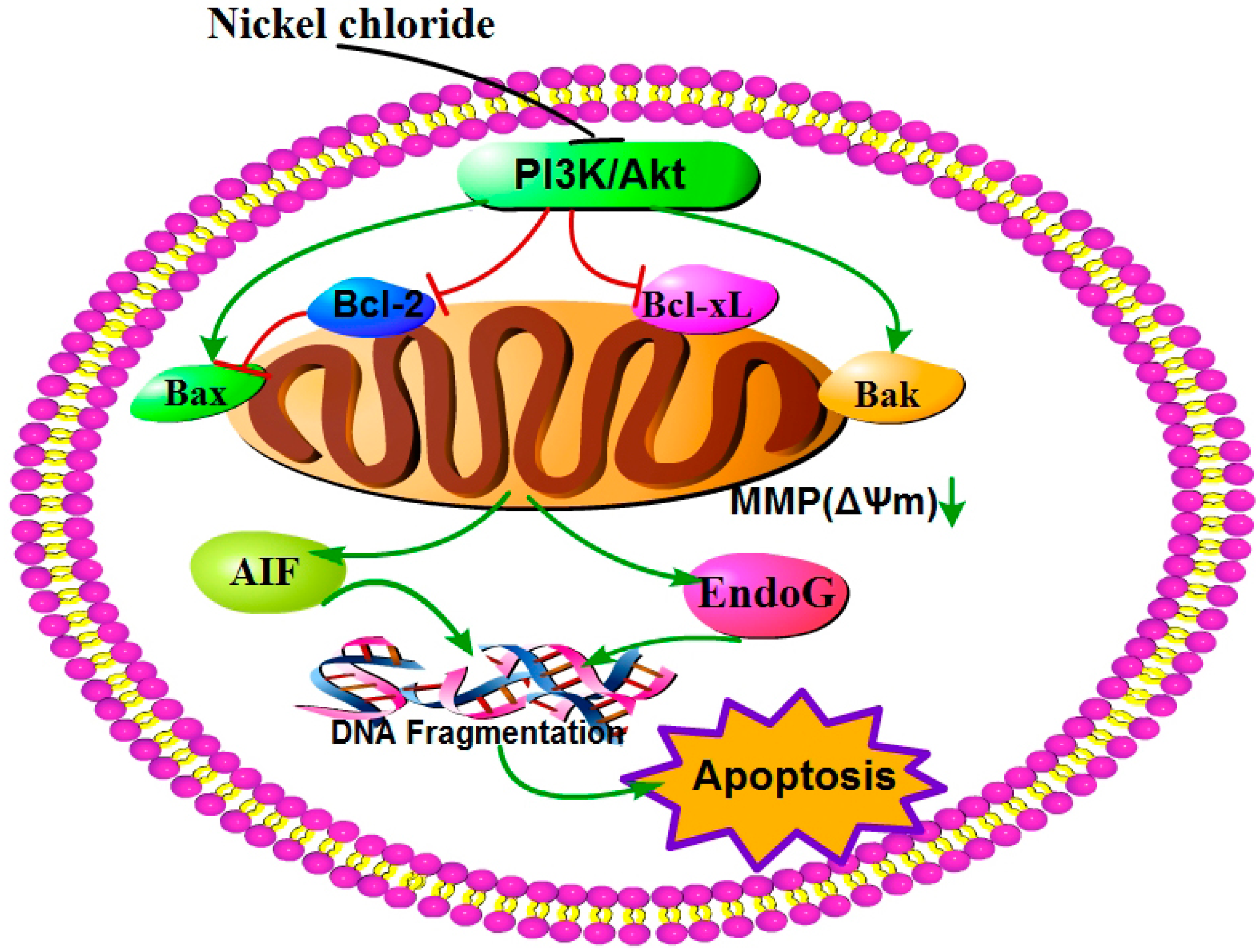
3. Discussion

4. Experimental Section
4.1. Animals and Treatment
4.2. Histopathological Examination of Kidney
4.3. Detection of Renal Apoptosis by TUNEL
4.4. Detection of Mitochondrial Membrane Potential (ΔѰm) in the Kidney by Flow Cytometry
4.5. Detection of Protein Expression in the Kidney by Immunohistochemistry
4.6. Detection of mRNA Expression in the Kidney by qRT-PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Accession Number | Primer | Primer Sequence (5ʹ–3ʹ) | Product Size (bp) | Tm (°C) |
|---|---|---|---|---|---|
| PI3K | NM001004410 | F | CGGATGTTGCCTTACGGTTGT | 162 | 58 |
| R | GTTCTTGTCCTTGAGCCACTGAT | ||||
| Akt1 | AF039943 | F | TGATGGCACATTCATTGGCTAC | 122 | 58 |
| R | TGTTTGGTTTAGGTCGTTCTGTCT | ||||
| Akt2 | AF181260 | F | CCGAAGTGCTGGAGGACAAC | 115 | 60 |
| R | CGCTCGTGGTCCTGGTTGTA | ||||
| Bcl-2 | NM205339 | F | GATGACCGAGTACCTGAACC | 114 | 61 |
| R | CAGGAGAAATCGAACAAAGGC | ||||
| Bax | XM422067 | F | TCCTCATCGCCATGCTCAT | 69 | 62 |
| R | CCTTGGTCTGGAAGCAGAAGA | ||||
| Bak | NM001030920 | F | TCTACCAGCAAGGCATCACGG | 122 | 60 |
| R | ATCGAGTGCAGCCACCCATC | ||||
| Bcl-xL | GU230783 | F | ATGAGTTTGAGCTGAGGTACCGG | 150 | 59 |
| R | AGAAGAAAGCCACGATGCGC | ||||
| AIF | NM001007490 | F | CTGGGTCCTGATGTGGGCTAT | 123 | 58 |
| R | TGTCCCTGACTGCTCTGTTGC | ||||
| EndoG | XM415487 | F | TGCCTGGAATAACCTTGAGAAATAC | 170 | 61 |
| R | TGAAGAAATGGGTAGGGACGG | ||||
| β-actin | L08165 | F | TGCTGTGTTCCCATCTATCG | 178 | 62 |
| R | TTGGTGACAATACCGTGTTCA |
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Poonkothai, M.; Vijayavathi, B.S. Nickel as an essential element and a toxicant. Int. J. Environ. Sci. 2012, 1, 285–288. [Google Scholar]
- Pasanen, K.; Pukkala, E.; Turunen, A.W.; Patama, T.; Jussila, I.; Makkonen, S.; Salonen, R.O.; Verkasalo, P.K. Mortality among population with exposure to industrial air pollution containing nickel and other toxic metals. J. Occup. Environ. Med. 2012, 54, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Cempel, M.; Nikel, G. Nickel: A review of its sources and environmental toxicology. Pol. J. Environ. Stud. 2006, 15, 375–382. [Google Scholar]
- Lu, H.; Shi, X.; Costa, M.; Huang, C. Carcinogenic effect of nickel compounds. Mol. Cell. Biochem. 2005, 279, 45–67. [Google Scholar] [CrossRef] [PubMed]
- Kubrak, O.I.; Husak, V.V.; Rovenko, B.M.; Poigner, H.; Kriews, M.; Abele, D.; Lushchak, V.I. Antioxidant system efficiently protects goldfish gills from Ni2+-induced oxidative stress. Chemosphere 2013, 90, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Alarifi, S.; Ali, D.; Alakhtani, S.; Al Suhaibani, E.S.; Al-Qahtani, A.A. Reactive oxygen species-mediated DNA damage and apoptosis in human skin epidermal cells after exposure to nickel nanoparticles. Biol. Trace Elem. Res. 2014, 157, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Das, K.K.; Das, S.N.; Dhundasi, S.A. Nickel, its adverse health effects & oxidative stress. Indian J. Med. Res. 2008, 128, 412–425. [Google Scholar] [PubMed]
- Uddin, A.N.; Burns, F.J.; Rossman, T.G.; Chen, H.; Kluz, T.; Costa, M. Dietary chromium and nickel enhance UV-carcinogenesis in skin of hairless mice. Toxicol. Appl. Pharm. 2007, 221, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Klein, C.B. Nickel carcinogenesis, mutation, epigenetics, or selection. Environ. Health Persp. 1999, 107, A438–A439. [Google Scholar] [CrossRef]
- Denkhausa E, S.K. Nickel essentiality, toxicity, and carcinogenicity. Crit. Rev. Oncol. Hematol. 2002, 42, 35–36. [Google Scholar] [CrossRef]
- Goodman, J.E.; Prueitt, R.L.; Dodge, D.G.; Thakali, S. Carcinogenicity assessment of water-soluble nickel compounds. Crit. Rev. Toxicol. 2009, 39, 365–417. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.H.; Liu, C.M.; Sun, J.M.; Feng, Z.J.; Cheng, C. Nickel-induced oxidative stress and apoptosis in Carassius auratus liver by JNK pathway. Aquat. Toxicol. 2014, 147, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Salnikow, K.; Costa, M. Epigenetic mechanisms of nickel carcinogenesis. J. Environ. Pathol. Toxicol. Oncol. 1999, 19, 307–318. [Google Scholar]
- Chen, C.-Y.; Lin, T.-K.; Chang, Y.-C.; Wang, Y.-F.; Shyu, H.-W.; Lin, K.-H.; Chou, M.-C. Nickel(II)-induced oxidative stress, apoptosis, G2/M arrest, and genotoxicity in normal rat kidney cells. J. Toxicol. Environ. Health A 2010, 73, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Şaplakoğlu, U.; İşcan, M.; İşcan, M. DNA single-strand breakage in rat lung, liver and kidney after single and combined treatments of nickel and cadmium. Mutat. Res. 1997, 394, 133–140. [Google Scholar] [CrossRef]
- Dally, H.; Hartwig, A. Induction and repair inhibition of oxidative DNA damage by nickel (II) and cadmium (II) in mammalian cells. Carcinogenesis 1997, 18, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Efremenko, A.Y.; Campbell, J.L., Jr.; Dodd, D.E.; Oller, A.R.; Clewell, H.J., 3rd. Time- and concentration-dependent genomic responses of the rat airway to inhaled nickel subsulfide. Toxicol. Appl. Pharmacol. 2014, 279, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Guo, H.; Deng, J.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Wang, X.; Wu, B.; Li, J.; Yin, S. Inhibitive effects of nickel chloride (NiCl2) on thymocytes. Biol. Trace Elem. Res. 2015, 164, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Huang, J. Toxicological effects of nickel chloride on the cytokine mRNA expression and protein levels in intestinal mucosal immunity of broilers. Environ. Toxicol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Huang, J. toxicological effects of nickel chloride on IgA+ B cells and sIgA, IgA, IgG, IgM in the intestinal mucosal immunity in broilers. Int. J. Environ. Res. Public Health 2014, 11, 8175–8192. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Huang, J. Dietary nickel chloride induces oxidative stress, apoptosis and alters Bax/Bcl-2 and caspase-3 mRNA expression in the cecal tonsil of broilers. Food Chem. Toxicol. 2014, 63, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Huang, J. Analysis of the Toll-like receptor 2-2 (TLR2-2) and TLR4 mRNA expression in the intestinal mucosal immunity of broilers fed on diets supplemented with nickel chloride. Int. J. Environ. Res. Public Health 2014, 11, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Li, J.; Yin, S.; Guo, H.; Deng, J.; Cui, H. Effects of nickel chloride on histopathological lesions and oxidative damage in the thymus. Health 2014, 6, 2875. [Google Scholar] [CrossRef]
- Huang, J.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Wu, B. Effect of dietary nickel chloride on splenic immune function in broilers. Biol. Trace Elem. Res. 2014, 159, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Wu, B. Downregulation of TLR4 and 7 mRNA expression levels in broiler’s spleen caused by diets supplemented with nickel chloride. Biol. Trace Elem. Res. 2014, 158, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Deng, J.; Yin, S.; Li, J.; Tang, K. NiCl2-down-regulated antioxidant enzyme mRNA expression causes oxidative damage in the broiler’s kidney. Biol. Trace Elem. Res. 2014, 162, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Huang, J.; Luo, Q.; Deng, Y.; Wang, H.; Liu, J. Changes of the serum cytokine contents in broilers fed on diets supplemented with nickel chloride. Biol. Trace Elem. Res. 2013, 151, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Huang, J. Dietary nickel chloride induces oxidative intestinal damage in broilers. Int. J. Environ. Res. Public Health 2013, 10, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Figlin, R.A. Phosphatidylinositol-3-kinase/Akt signaling pathway and kidney cancer, and the therapeutic potential of phosphatidylinositol-3-kinase/Akt inhibitors. J. Urol. 2009, 182, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Ma, J.Q.; Sun, Y.Z. Puerarin protects rat kidney from lead-induced apoptosis by modulating the PI3K/Akt/eNOS pathway. Toxicol. Appl. Pharmacol. 2012, 258, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–41. [Google Scholar] [CrossRef]
- Harris, G.K.; Shi, X. Signaling by carcinogenic metals and metal-induced reactive oxygen species. Mutat. Res. 2003, 533, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.V. Metals and apoptosis: Recent developments. J. Trace Elem. Med. Biol. 2008, 22, 262–84. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.C.; Youle, R.J. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. Bcl-2 family members and the mitochondria in apoptosis. Gene Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Shyu, H.W.; Chang, Y.C.; Tseng, W.C.; Huang, Y.L.; Lin, K.H.; Chou, M.C.; Liu, H.L.; Chen, C.Y. Nickel (II)-induced cytotoxicity and apoptosis in human proximal tubule cells through a ROS- and mitochondria-mediated pathway. Toxicol. Appl. Pharmacol. 2012, 259, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wu, B. The association between splenocyte apoptosis and alterations of Bax, Bcl-2 and Caspase-3 mRNA expression, and oxidative stress induced by dietary nickel chloride in broilers. Int. J. Environ. Res. Public Health 2013, 10, 7310–7326. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Song, M.; Zhang, Y.; Yan, M.; Zhang, M.; Bi, H. Nickel nanowires induce cell cycle arrest and apoptosis by generation of reactive oxygen species in HeLa cells. Toxicol. Rep. 2014, 1, 114–121. [Google Scholar] [CrossRef]
- Hossain, M.Z.; Kleve, M.G. Nickel nanowires induced and reactive oxygen species mediated apoptosis in human pancreatic adenocarcinoma cells. Int. J. Nanomed. 2011, 6, 1475. [Google Scholar] [CrossRef] [PubMed]
- Gathwan, K.H.; Al-Karkhi, I.H.T.; Jaffar al-Mulla, E.A. Hepatic toxicity of nickel chloride in mice. Res. Chem. Intermed. 2012, 39, 2537–2542. [Google Scholar] [CrossRef]
- Vaux, D.L. Apoptogenic factors released from mitochondria. Biochim. Biophys. Acta 2011, 1813, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Zamzami, N.; Castedo, M.; Hirsch, T.; Marchetti, P.; Macho, A.; Daugas, E.; Geuskens, M.; Kroemer, G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 1996, 184, 1331–41. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Wang, X.L.X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bowman, L.; Zhang, X.; Shi, X.; Jiang, B.; Castranova, V.; Ding, M. Metallic nickel nano- and fine particles induce JB6 cell apoptosis through a caspase-8/AIF mediated cytochrome c-independent pathway. J. Nanobiotechnol. 2009, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 2009, 122, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, J.P.; Weller, M. Apoptosis in gliomas: Molecular mechanisms and therapeutic implications. J. Neurooncol. 2004, 70, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Korsmeyer, S.J. BCL-2 gene family and the regulation of programmed cell death. Cancer Res. 1999, 59, 1693s–1700s. [Google Scholar] [CrossRef]
- Osaki, M.; Oshimura, M.A.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Nihal, M.; Fu, V.X.; Jarrard, D.F.; Ahmad, N. Resveratrol-caused apoptosis of human prostate carcinoma LNCaP cells is mediated via modulation of phosphatidylinositol 3ʹ-kinase/Akt pathway and Bcl-2 family proteins. Mol. Cancer Ther. 2006, 5, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Sun, Y.; Hu, J. Catalpol inhibits apoptosis in hydrogen peroxide-induced endothelium by activating the PI3K/Akt signaling pathway and modulating expression of Bcl-2 and Bax. Eur. J. Pharmacol. 2010, 628, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.J.; Chang, Q.S.; Wang, X.; Son, Y.O.; Liu, J.; Zhang, Z.; Bi, Y.Y.; Shi, X. Activation of Akt/GSK3beta and Akt/Bcl-2 signaling pathways in nickel-transformed BEAS-2B cells. Int. J. Oncol. 2011, 39, 1285–1294. [Google Scholar] [PubMed]
- Liu, C.M.; Zheng, G.H.; Ming, Q.L.; Chao, C.; Sun, J.M. Sesamin protects mouse liver against nickel-induced oxidative DNA damage and apoptosis by the PI3K-Akt pathway. J. Agric. Food Chem. 2013, 61, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Chang, Q.; Wang, X.; Son, Y.; Zhang, Z.; Chen, G.; Luo, J.; Bi, Y.; Chen, F.; Shi, X. Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cells. Chem. Res. Toxicol. 2010, 23, 568–577. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (NRC). Nutrient Requirements of Poultry, 9th ed.; National Academy Press: Washington, DC, USA, 1994. [Google Scholar]
- Ling, J.; Leach, R. Studies on nickel metabolism: Interaction with other mineral elements. Poult. Sci. 1979, 58, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.W.; Reid, B.L. Nickel toxicity in growing chicks. J. Nutr. 1968, 95, 612–616. [Google Scholar] [PubMed]
- Szilagyi, M.; Szentmihalyi, S.; Anke, M. Changes in some of the biochemical parameters in Ni and Mo deficient animals [goat, sheep, pig, chicken, rat]. Proc. (Hungary). 1981, Volume 1, pp. 257–260, International System for Agricultural Science and Technology. Available online: http://agris.fao.org/agris-search/search.do?recordID=HU8200908 (accessed on 24 April 2015).
- Bersényi, A.; Fekete, S.G.; Szilágyi, M.; Berta, E.; Zöldág, L.; Glávits, R. Effects of nickel supply on the fattening performance and several biochemical parameters of broiler chickens and rabbits. Acta Vet. Hung. 2004, 52, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Wu, B.; Chen, K.; Deng, J. Modulation of the PI3K/Akt Pathway and Bcl-2 Family Proteins Involved in Chicken’s Tubular Apoptosis Induced by Nickel Chloride (NiCl2). Int. J. Mol. Sci. 2015, 16, 22989-23011. https://doi.org/10.3390/ijms160922989
Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K, Deng J. Modulation of the PI3K/Akt Pathway and Bcl-2 Family Proteins Involved in Chicken’s Tubular Apoptosis Induced by Nickel Chloride (NiCl2). International Journal of Molecular Sciences. 2015; 16(9):22989-23011. https://doi.org/10.3390/ijms160922989
Chicago/Turabian StyleGuo, Hongrui, Hengmin Cui, Xi Peng, Jing Fang, Zhicai Zuo, Junliang Deng, Xun Wang, Bangyuan Wu, Kejie Chen, and Jie Deng. 2015. "Modulation of the PI3K/Akt Pathway and Bcl-2 Family Proteins Involved in Chicken’s Tubular Apoptosis Induced by Nickel Chloride (NiCl2)" International Journal of Molecular Sciences 16, no. 9: 22989-23011. https://doi.org/10.3390/ijms160922989
APA StyleGuo, H., Cui, H., Peng, X., Fang, J., Zuo, Z., Deng, J., Wang, X., Wu, B., Chen, K., & Deng, J. (2015). Modulation of the PI3K/Akt Pathway and Bcl-2 Family Proteins Involved in Chicken’s Tubular Apoptosis Induced by Nickel Chloride (NiCl2). International Journal of Molecular Sciences, 16(9), 22989-23011. https://doi.org/10.3390/ijms160922989