
Enzymatic Kinetic Resolution of 2-Piperidineethanol for the Enantioselective Targeted and Diversity Oriented Synthesis †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Enantiopure 2-Piperidineethanol: Resolution versus Synthesis
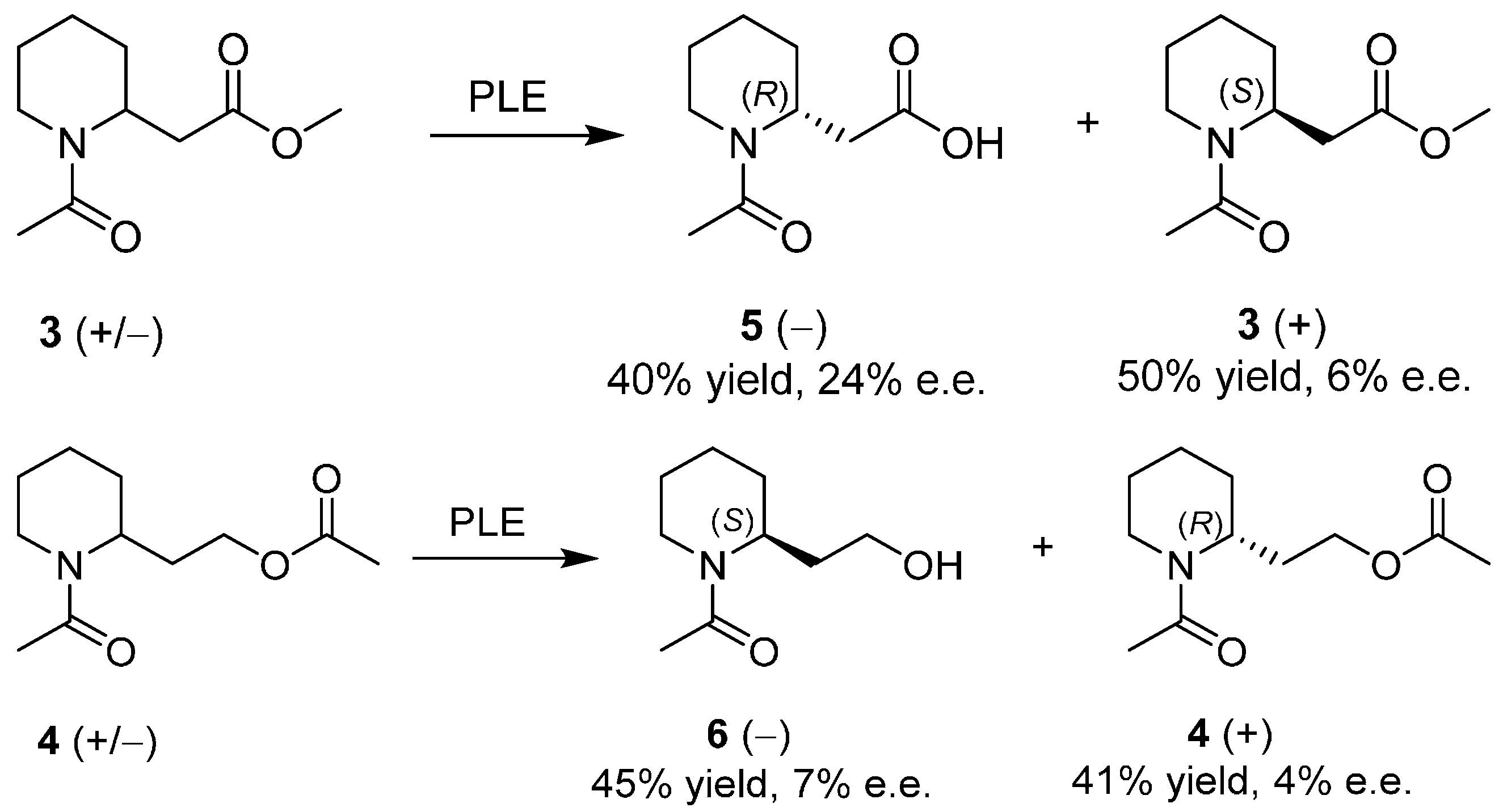
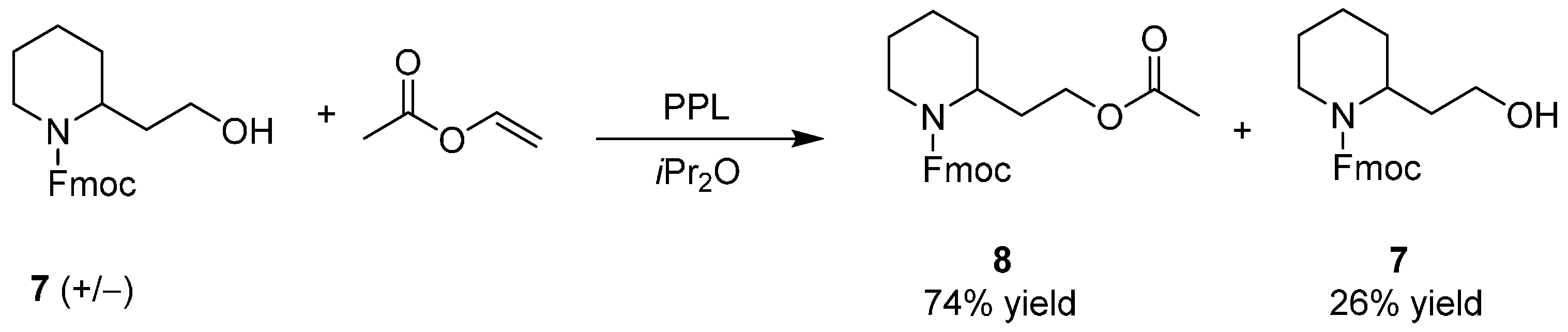
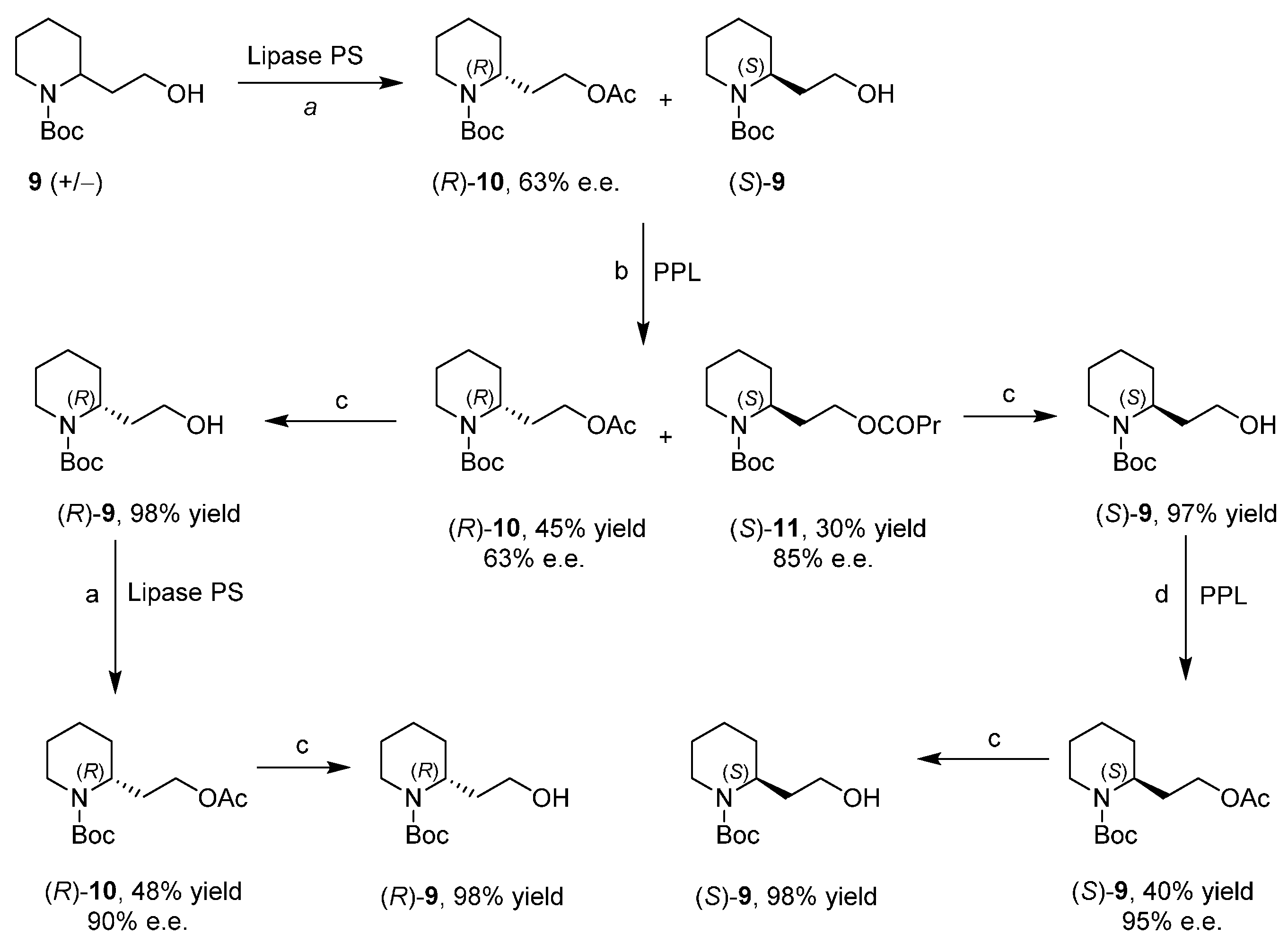
Enantiopure 2-Piperidineethanol by Enzymatic Kinetic Resolutions





3. An Overview of the Synthetic Strategies in the Synthesis of Alkaloids Starting from Either Enantiopure 1 or 2

3.1. Enantioselective Synthesis of the Alkaloid (+)-Aloperine


3.2. Enantioselective Synthesis of the Alkaloid Boehmeriasin A

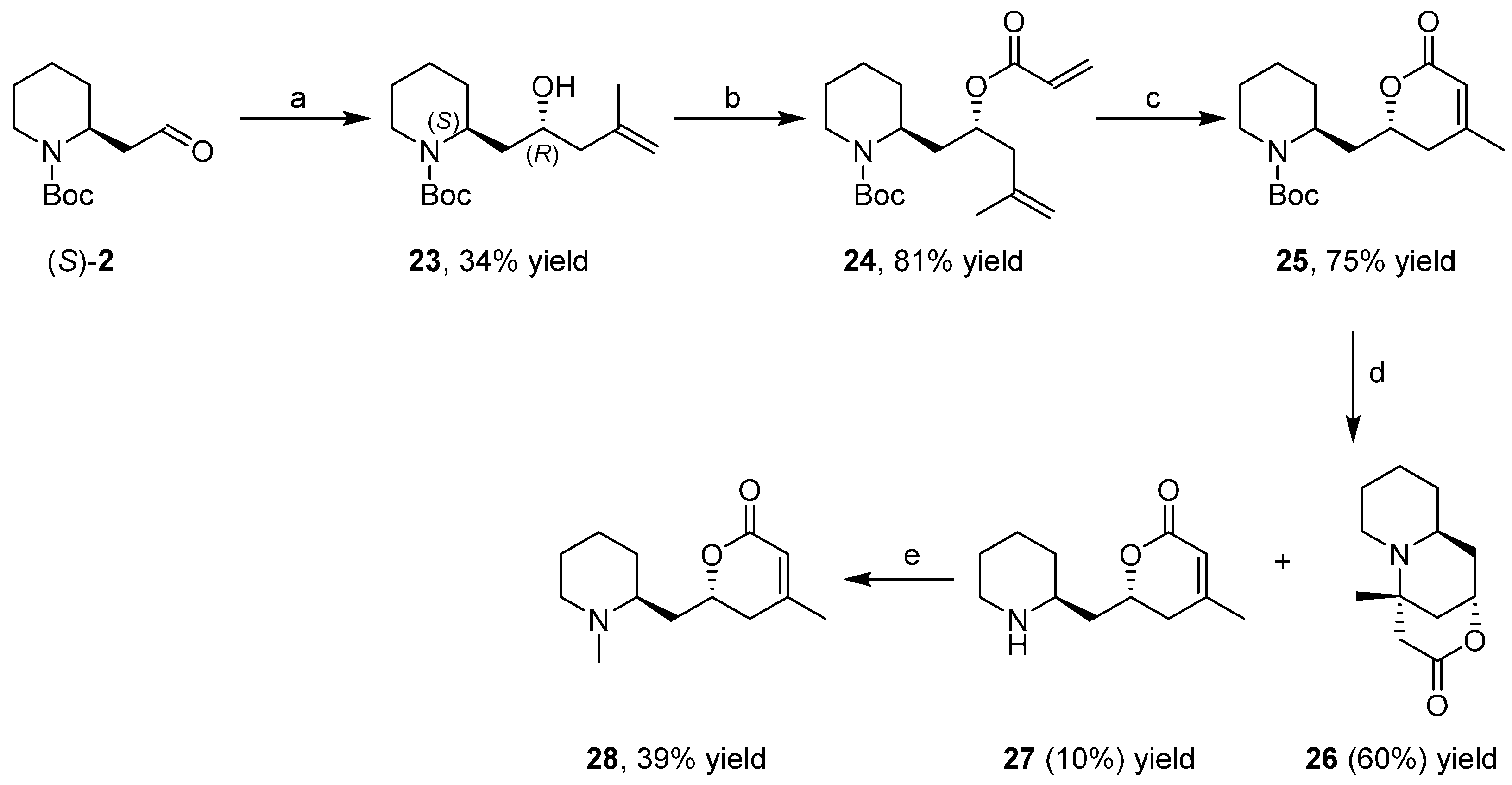
3.3. Enantioselective Synthesis of the Alkaloid Dumetorine

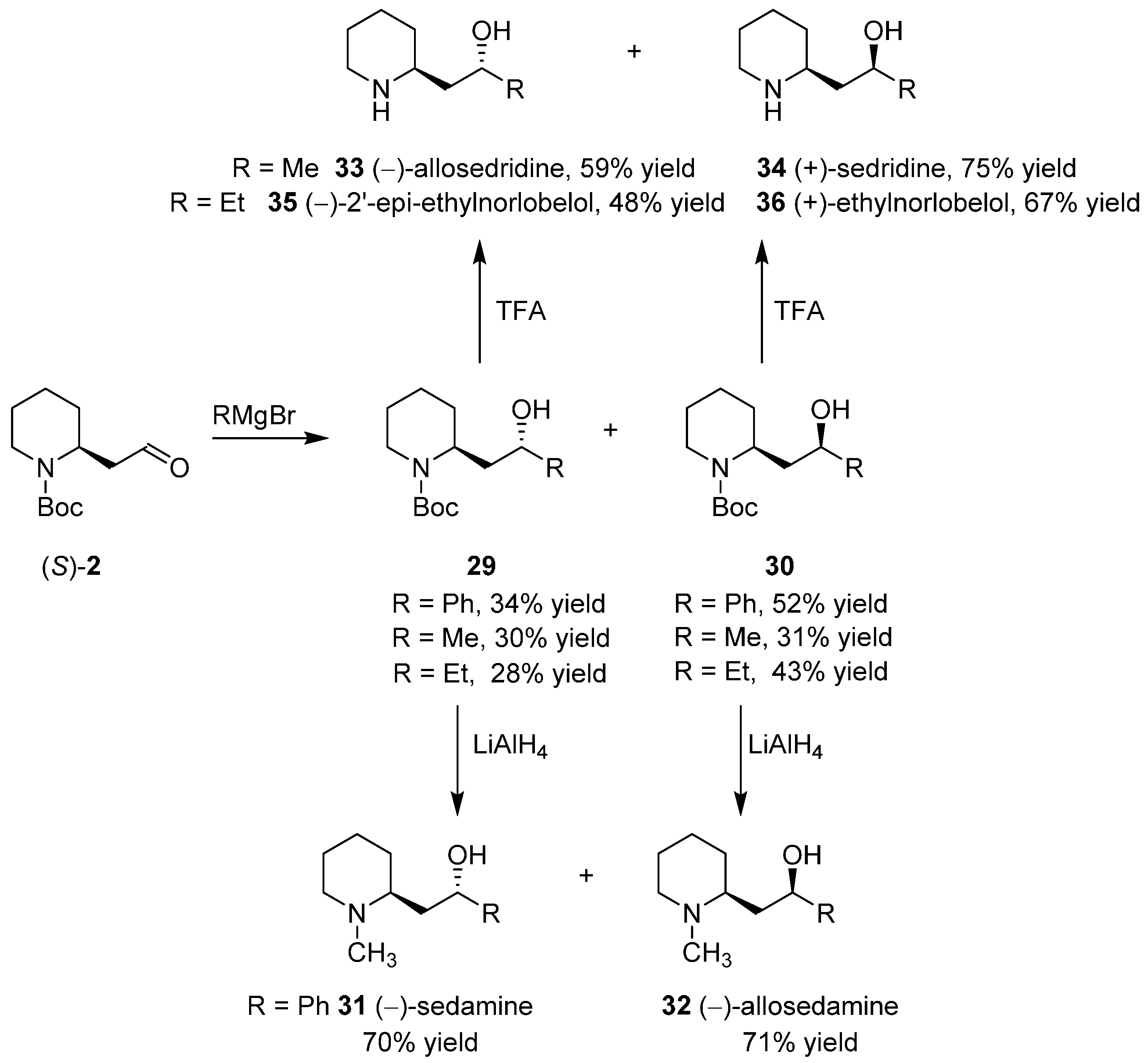
3.4. Enantioselective Synthesis of the Alkaloids Sedamine, Allosedamine, Sedridines and Ethylnorlobelols

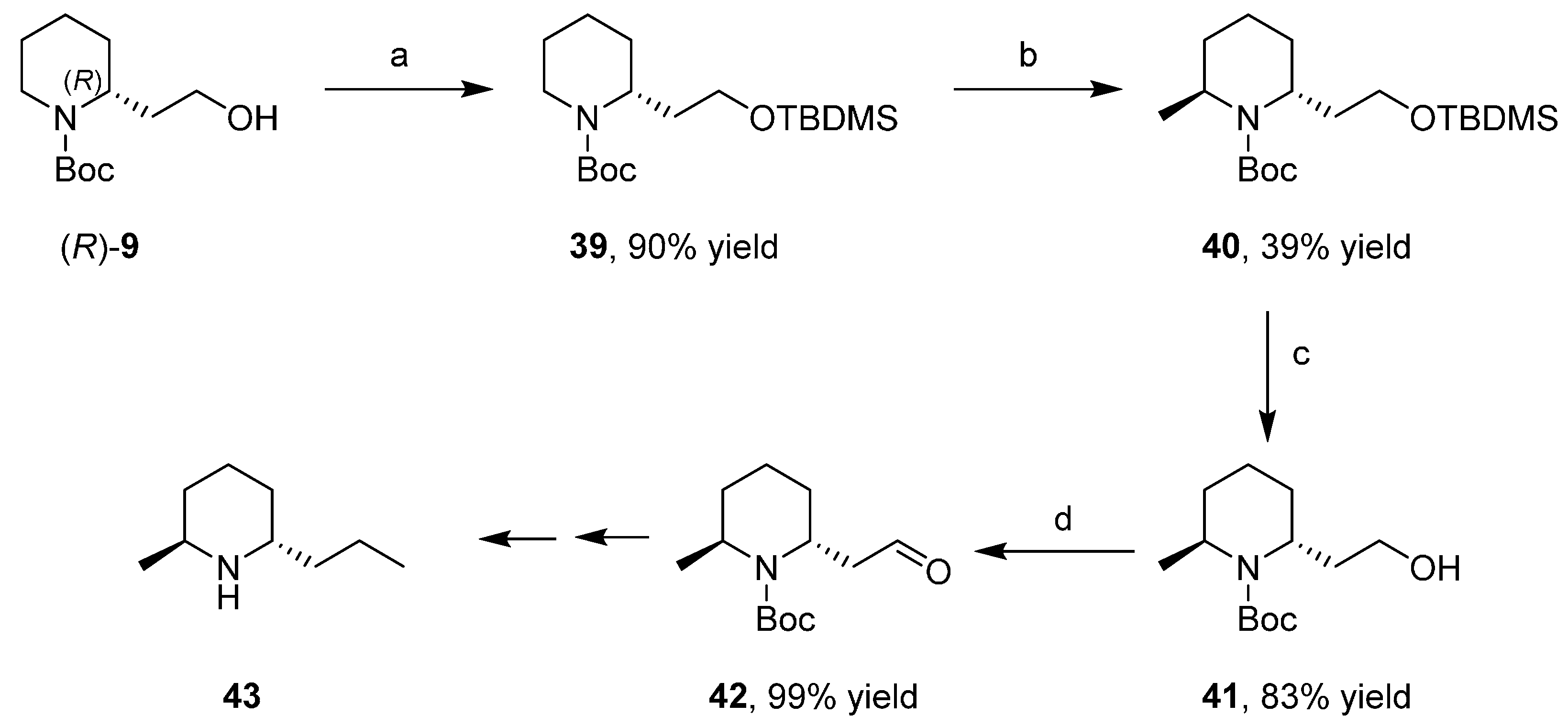
3.5. Synthesis of the Enantiopure Alkaloids Coniine and Epidihydropinidine


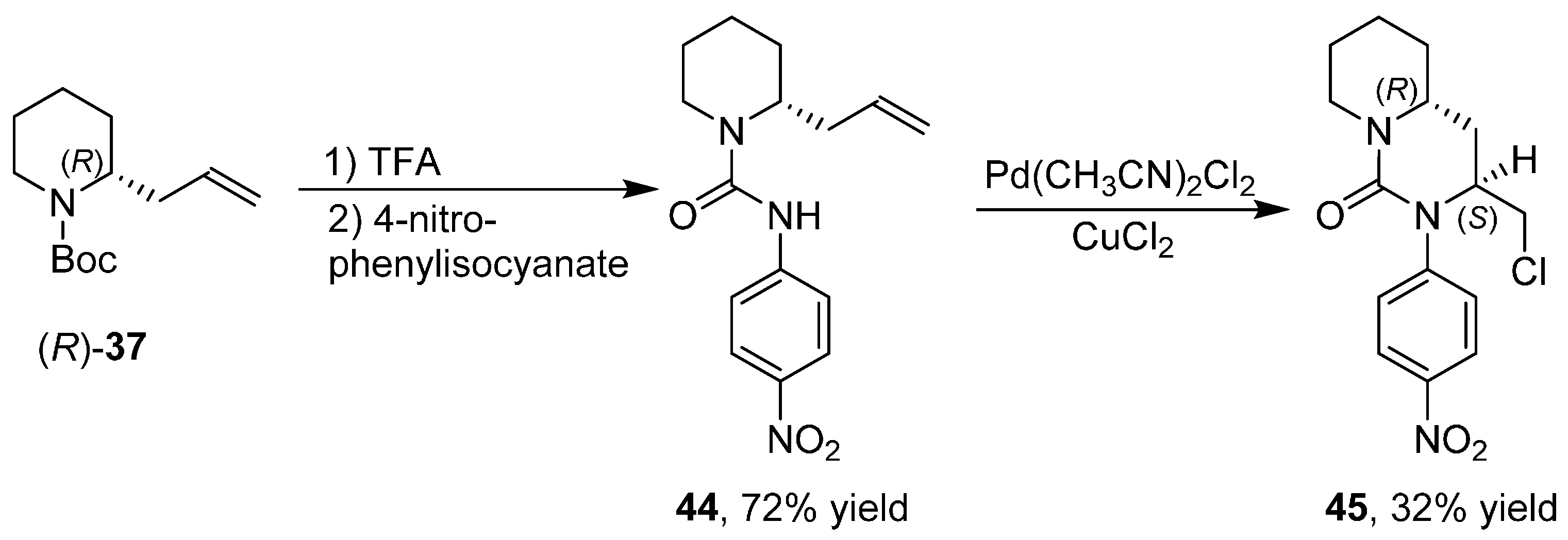
3.6. Enantioselective Synthesis of Polycyclic Piperidine Derivatives



4. A General Overview of the Structures of Alkaloids Synthesized from both 1 or 2
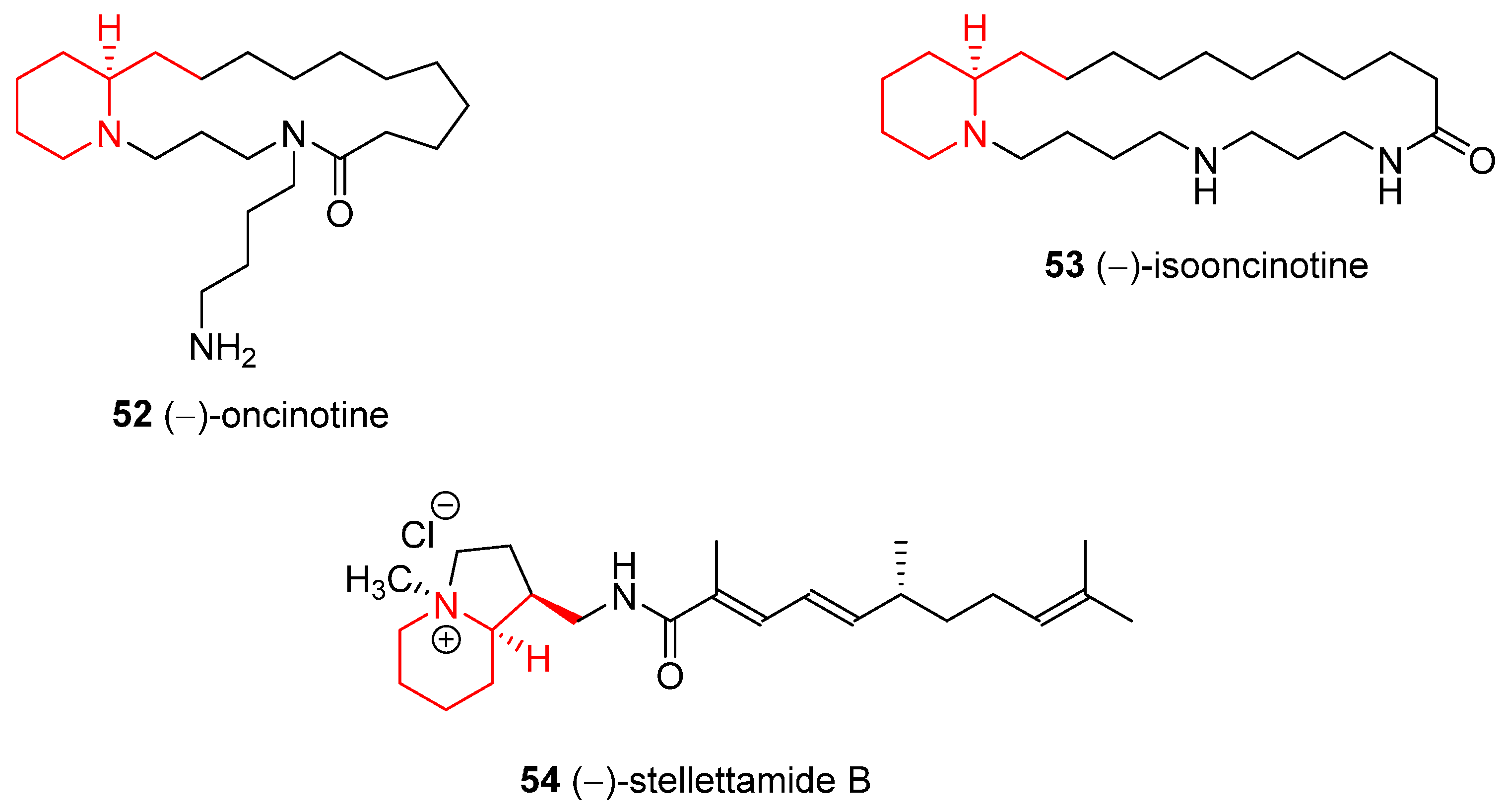
4.1. Enantioselective Synthesis of the Alkaloids (−)-Oncinotine, (−)-Isooncinotine, and (−)-Stellettamide B

4.2. Enantioselective Synthesis of the Alkaloids (+)-Vertine and (+)-Lythrine

4.3. Enantioselective Synthesis of the Alkaloids (+)-Myrtine, (−)-Lupinine, and (+)-Epiepiquinamide

4.4. Enantioselective Synthesis of the Alkaloids (−)-Cermizine C, (−)-Senepodine G and (+)-Cermizine D

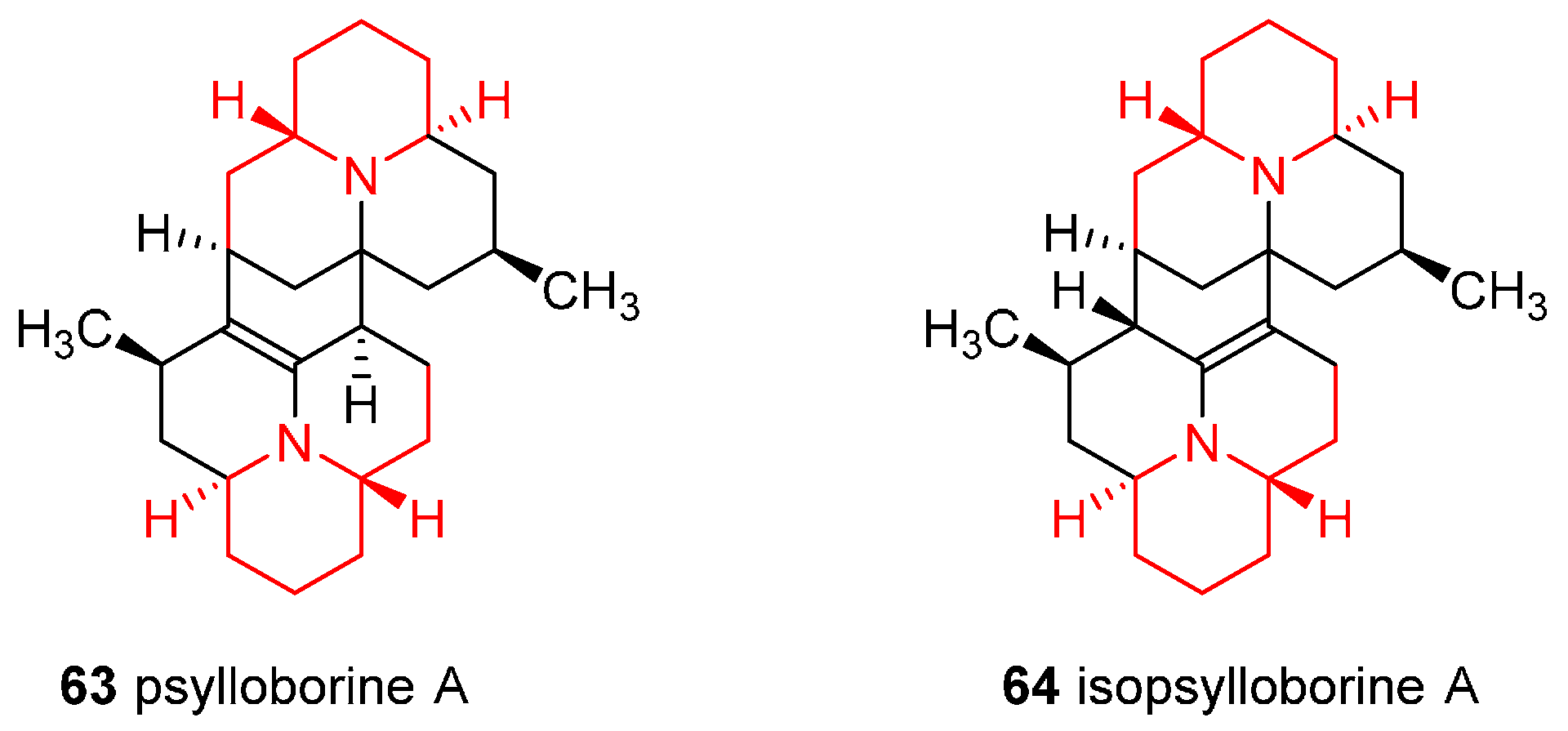
4.5. Enantioselective Synthesis of the Alkaloids Psylloborine A and Isopsylloborine A

4.6. Enantioselective Synthesis of the Alkaloids Tetraponerines T3, T4, T7, and T8

4.7. Enantioselective Synthesis of the Alkaloids (+)-Conhydrine and 4-(2-(Piperidin-2-yl)ethoxy)quinoline Derivatives

5. Conclusions
Author Contributions
Conflicts of Interest
References
- Freifelder, M.; Robinson, R.M.; Stone, G.R. Hydrogenation of substituted pyridines with rhodium on carbon catalyst. J. Org. Chem. 1962, 27, 284–286. [Google Scholar] [CrossRef]
- Paul, S.; Ghoshal, A.K.; Mandal, B. Absorption of carbon dioxide into aqueous solutions of 2-piperidineethanol: kinetics analysis. Ind. Eng. Chem. Res. 2009, 48, 1414–1419. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, Y. Cyclic Organosilicon Compounds as Electron Donors in Ziegler-Natta Catalyst Systems for Producing Propylene Polymer Having High Melt-Flowability. PCT Int. Appl. Patent WO 2013063464, 2 May 2013. [Google Scholar]
- Krueger, B.W.; Sasse, K.; Hoever, F.P.; Nentwig, G.; Behrenz, W. Preparation of α,ω-Amino Alcohols as Insect and Mite Repellents. Eur. Appl. Patent EP 289842, 9 November 1988. [Google Scholar]
- He, H.; Zhao, D.; Zhu, Y. Process for Preparation of Insect Repellent Icaridin. Chinese Patent CN 102167681, 31 August 2011. [Google Scholar]
- Harder, A.; von Samson-Himmelstjerna, G.; Kruger, B.W.; Mehlhorn, H.; Schmidt, J. Aminocarbonyloxy Compound Anthelminthic Agents for the Prevention of Parasitic Infections in Humans and Animals. PCT Int. Appl. Patent WO 2002002115, 10 January 2002. [Google Scholar]
- Gaillard, P.; Johnson, C.N.; Novelli, R.; Porter, R.A.; Stemp, G.; Thewlis, K.M. Preparation of N-Aroyl Cyclic Amines as Orexin Receptor Antagonists. PCT Int. Appl. Patent WO 2002089800, 14 November 2002. [Google Scholar]
- Carson, J.R.; Carmosin, R.J.; Vaught, J.L.; Gardocki, J.F.; Costanzo, M.J.; Raffa, R.B.; Almond, H.R., Jr. 2-Substituted 1-azabicycloalkanes, a new class of non-opiate antinociceptive agents. J. Med. Chem. 1992, 35, 2855–2863. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.H.; Copper, D.G. Preparation of (Aryloxyalkyl)piperidines and Analogs as Calcium Channel Antagonists. PCT Int. Appl. Patent WO 9533723, 14 December 1995. [Google Scholar]
- Forbes, I.T.; Rahman, S.K. Preparation of Sulfonamide Derivatives as 5-HT7 Receptor Antagonists . PCT Int. Appl. Patent WO 9749695, 31 December 1997. [Google Scholar]
- Thompson, S.K.; Priestley, T.; Smith, R.A.; Saha, A.K.; Rudra, S.; Hajra, A.K.; Chatterjee, D.; Behrens, C.H.; He, Y.; Li, H.Y. Aminoindane Compounds as Sodium Channel Inhibitors and TRPV1 Receptor Activator and Their Preparation and Use in the Treatment of Pain. PCT Int. Appl. Patent WO 2012112969, 23 August 2012. [Google Scholar]
- Zanka, A.; Uematsu, R.; Morinaga, Y.; Yasuda, H.; Yamazaki, H. Process development of a novel non-xanthine adenosine A1 receptor antagonist. Org. Process. Res. Dev. 1999, 3, 389–393. [Google Scholar] [CrossRef]
- Aberg, G.; Chen, J.L. The R-Isomer of 2-[2-(N-(2-Indanyl)-N-phenylamino)ethyl]piperidine and Other Dermal Anesthetic Agents for Inducing Local Anesthesia, Analgesia and Sleep. PCT Int. Appl. Patent WO 2006037047, 6 April 2006. [Google Scholar]
- Young, J.R.; Huang, S.X.; Chen, I.; Walsh, T.F.; de Vita, R.J.; Wyvratt, M.J.; Goulet, M.T.; Ren, N.; Lo, J.; Yang, Y.T.; et al. Quinolones as gonadotropin releasing hormone (GnRH) antagonists: Simultaneous optimization of the C(3)-aryl and C(6)-substituents. Bioorg. Med. Chem. Lett. 2000, 10, 1723–1727. [Google Scholar] [CrossRef]
- Paruch, K.; Dwyer, M.P.; Alvarez, C.; Brown, C.; Chan, T.Y.; Doll, R.J.; Keertikar, K.; Knutson, C.; McKittrick, B.; Rivera, J.; et al. Discovery of Dinaciclib (SCH 727965): A potent and selective inhibitor of cyclin-dependent kinases. ACS Med. Chem. Lett. 2010, 1, 204–208. [Google Scholar] [CrossRef] [PubMed]
- DeVita, R.J.; Goulet, M.T.; Wyvratt, M.J.; Fisher, M.H.; Lo, J.L.; Yang, Y.T.; Cheng, K.; Smith, R.G. Investigation of the 4-O-alkylamine substituent of non-peptide quinolone GnRH receptor antagonists. Bioorg. Med. Chem. Lett. 1999, 9, 2621–2624. [Google Scholar] [CrossRef]
- Danilewicz, J.C.; Abel, S.M.; Brown, A.D.; Fish, P.V.; Hawkeswood, E.; Holland, S.J.; James, K.; McElroy, A.B.; Overington, J.; Powling, M.J.; et al. Design of selective thrombin inhibitors based on the (R)-Phe-Pro-Arg sequence. J. Med. Chem. 2002, 45, 2432–2453. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Maeda, M.; Kibayashi, C. Diastereofacial selectivity in intermolecular nitrone cycloadditions to chiral allyl ethers. Application to chiral synthesis of coniine. Tetrahedron Lett. 1992, 33, 3765–3768. [Google Scholar]
- Ina, H.; Ito, M.; Kibayashi, C. Enantioselective Total synthesis of the macrocyclic spermidine alkaloid (−)-Oncinotine. J. Org. Chem. 1996, 61, 1023–1029. [Google Scholar] [CrossRef]
- Yamazaki, N.; Dokoshi, W.; Kibayashi, C. Total synthesis of (−)-stellettamide b and determination of its absolute stereochemistry. Org. Lett. 2001, 3, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Bosque, I.; Gonzalez-Gomez, J.C.; Guijarro, A.; Foubelo, F.; Yus, M. Concise total synthesis and stereochemical analysis of Tetraponerines T3 and T4. J. Org. Chem. 2012, 77, 10340–10346. [Google Scholar] [CrossRef] [PubMed]
- Satyalakshmi, G.; Suneel, K.; Shinde, D.B.; Das, B. Stereoselective total synthesis of the piperidine alkaloids, (+)-coniine, (+)-pseudoconhydrine, and (+)-sedamine through a common intermediate. Tetrahedron Asymmetry 2011, 22, 1000–1005. [Google Scholar] [CrossRef]
- Fustero, S.; Jimenez, D.; Moscardo, J.; Catalan, S.; del Pozo, C. Enantioselective organocatalytic intramolecular aza-michael reaction: A concise synthesis of (+)-Sedamine, (+)-Allosedamine, and (+)-Coniine. Org. Lett. 2007, 9, 5283–5286. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.C.; Rathbone, L.K.; Yang, H.; Collett, N.D.; Carter, R.G. Improved protocol for asymmetric, intramolecular heteroatom michael addition using organocatalysis: Enantioselective syntheses of homoproline, pelletierine, and homopipecolic acid. J. Org. Chem. 2008, 73, 5155–5158. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Panda, G. l-Proline derived nitrogenous steroidal systems: An asymmetric approach to 14-aza steroids. RSC Adv. 2013, 3, 19533–19544. [Google Scholar] [CrossRef]
- Toy, M.S.; Price, C.C. d- and l-Polyconidine. J. Am. Chem. Soc. 1960, 82, 2613–2616. [Google Scholar] [CrossRef]
- Callens, R.; Gire, R.; Pousset, C. Use of Enantiopure N-Sulfonylpyroglutamic Acids as Resolving Agents for Amines and Amino Alcohols. PCT Int. Appl. Patent WO 2009080708, 2 July 2009. [Google Scholar]
- Chen, F.X.; Tamarez, M.M.; Xie, J. Process for Resolving (S)-2-Piperidin-2-ylethanol from a Mixture of Isomers Using N-Acetyl-l-leucine or N-Acetyl-l-methionine. PCT Int. Appl. Patent WO 2008021509, 21 February 2008. [Google Scholar]
- Koeller, K.M.; Wong, C.H. Enzymes for chemical synthesis. Nature 2001, 409, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Carrea, G.; Riva, S. Properties and synthetic applications of enzymes in organic solvents. Angew. Chem. Int. Ed. 2000, 39, 2226–2254. [Google Scholar] [CrossRef]
- Santaniello, E.; Ferraboschi, P.; Grisenti, P.; Manzocchi, A. The biocatalytic approach to the preparation of enantiomerically pure chiral building blocks. Chem. Rev. 1992, 92, 1071–1140. [Google Scholar] [CrossRef]
- Toone, E.J.; Jones, J.B. Enzymes in organic synthesis. 40. Evaluation of the enantioselectivity of the pig liver esterase-catalyzed hydrolyses of racemic piperidine carboxylic acid esters. Can. J. Chem. 1987, 65, 2722–2726. [Google Scholar] [CrossRef]
- Eguchi, T.; Mochida, K. Manufacture of Optically Active Piperidines with Lipase. Japanese Patent JP 06014798, 25 January 1994. [Google Scholar]
- Angoli, M.; Barilli, A.; Lesma, G.; Passarella, D.; Riva, S.; Silvani, A.; Danieli, B. Remote stereocenter discrimination in the enzymatic resolution of piperidine-2-ethanol. short enantioselective synthesis of sedamine and allosedamine. J. Org. Chem. 2003, 68, 9525–9527. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.M. Improving enzymes by using them in organic solvents. Nature 2001, 409, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Belinghieri, F.; Passarella, D.; Lesma, G.; Riva, S.; Silvani, A.; Danieli, B. Enzyme assisted enantioselective synthesis of the alkaloid (+)-aloperine. Tetrahedron Asymmetry 2004, 15, 2921–2925. [Google Scholar] [CrossRef]
- Passarella, D.; Angoli, M.; Giardini, A.; Lesma, G.; Silvani, A.; Danieli, B. Concise total synthesis of (±)-aloperine and 6-epi-aloperine. Org. Lett. 2002, 4, 2925–2928. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, M.S.; Calogero, F.; Baumann, M.; Garcia-Argaez, A.N.; Pieraccini, S.; Sironi, M.; Dapiaggi, F.; Bucci, R.; Broggini, G.; Gazzola, S.; et al. Boehmeriasin A as new lead compound for the inhibition of topoisomerases and SIRT2. Eur. J. Med. Chem. 2015, 92, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Song, J.; Lee, H.; Kim, E.-Y.; Lee, K.; Lee, S.K.; Kim, S. Design, synthesis, and biological activity of sulfonamide analogues of Antofine and Cryptopleurine as potent and orally active antitumor agents. J. Med. Chem. 2015, 58, 7749–7762. [Google Scholar] [CrossRef] [PubMed]
- Passarella, D.; Riva, S.; Grieco, G.; Cavallo, F.; Checa, B.; Arioli, F.; Riva, E.; Comi, D.; Danieli, B. Enantiopure N-Boc piperidine-2-ethanol for the synthesis of (+)- and (−)-dumetorine, and (+)- and (−)-epidihydropinidine. Tetrahedron Asymmetry 2009, 20, 192–197. [Google Scholar] [CrossRef]
- Riva, E.; Rencurosi, A.; Gagliardi, S.; Passarella, D.; Martinelli, M. Synthesis of (+)-Dumetorine and Congeners by Using Flow Chemistry Technologies. Chem. Eur. J. 2011, 17, 6221–6226. [Google Scholar] [CrossRef] [PubMed]
- Passarella, D.; Barilli, A.; Belinghieri, F.; Fassi, P.; Riva, S.; Sacchetti, A.; Silvani, A.; Danieli, B. Short enantioselective synthesis of sedridines, ethylnorlobelols and coniine via reagent-based differentiation. Tetrahedron Asymmetry 2005, 16, 2225–2229. [Google Scholar] [CrossRef]
- Borsini, E.; Broggini, G.; Colombo, F.; Khansaa, M.; Fasana, A.; Galli, S.; Passarella, D.; Riva, E.; Riva, S. Enantiopure 2-piperidylacetaldehyde as a useful building block in the diversity-oriented synthesis of polycyclic piperidine derivatives. Tetrahedron Asymmetry 2011, 22, 264–269. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Hou, D.R. Efficient, asymmetric synthesis of (−)-isooncinotine. Tetrahedron 2007, 63, 3000–3005. [Google Scholar] [CrossRef]
- Chausset-Boissarie, L.; Arvai, R.; Cumming, G.R.; Guenee, L.; Kundig, E.P. Asymmetric synthesis of (+)-vertine and (+)-lythrine. Org. Biomol. Chem. 2012, 10, 6473–6479. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Moscardo, J.; Sanchez-Rosello, M.; Flores, S.; Guerola, M.; del Pozo, C. Organocatalytic enantioselective synthesis of quinolizidine alkaloids (+)-myrtine, (−)-lupinine, and (+)-epiepiquinamide. Tetrahedron 2011, 67, 7412–7417. [Google Scholar] [CrossRef]
- Snider, B.B.; Grabowski, J.F. Total synthesis of (−)-Senepodine G and (−)-Cermizine C. J. Org. Chem. 2007, 72, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Veerasamy, N.; Carlson, E.C.; Collett, N.D.; Saha, M.; Carter, R.G. Enantioselective approach to quinolizidines: Total synthesis of Cermizine D and formal syntheses of Senepodine G and Cermizine C. J. Org. Chem. 2013, 78, 4779–4800. [Google Scholar] [CrossRef] [PubMed]
- Veerasamy, N.; Carlson, E.C.; Carter, R.G. Expedient enantioselective synthesis of Cermizine D. Org. Lett. 2012, 14, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, T.C.; Trotta, A.H.; Snyder, S.A. A Strategy for complex dimer formation when biomimicry fails: Total synthesis of ten coccinellid alkaloids. J. Am. Chem. Soc. 2014, 136, 9743–9753. [Google Scholar] [CrossRef] [PubMed]
- Bosque, I.; Gonzalez-Gomez, J.C.; Loza, M.I.; Brea, J. Natural tetraponerines: A general synthesis and antiproliferative activity. J. Org. Chem. 2014, 79, 3982–3991. [Google Scholar] [CrossRef] [PubMed]
- Airiau, E.; Girard, N.; Pizzeti, M.; Salvadori, J.; Taddei, M.; André, M. Hydroformylation of alkenylamines. Concise approaches toward piperidines, quinolizidines, and related alkaloids. J. Org. Chem. 2010, 75, 8670–8673. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.M.; Sudalai, A. Enantioselective synthesis of (+)-α-Conhydrine and (–)-Sedamine by l-Proline-Catalysed α-Aminooxylation. Eur. J. Org. Chem. 2010, 3437–3444. [Google Scholar] [CrossRef]
- Wolkenberg, S.E.; Zhao, Z.; Thut, C.; Maxwell, J.W.; McDonald, T.P.; Kinose, F.; Reilly, M.; Lindsley, C.W.; Hartman, G.D. Design, synthesis, and evaluation of novel 3,6-diaryl-4-aminoalkoxyquinolines as selective agonists of somatostatin receptor subtype 2. J. Med. Chem. 2011, 54, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perdicchia, D.; Christodoulou, M.S.; Fumagalli, G.; Calogero, F.; Marucci, C.; Passarella, D. Enzymatic Kinetic Resolution of 2-Piperidineethanol for the Enantioselective Targeted and Diversity Oriented Synthesis. Int. J. Mol. Sci. 2016, 17, 17. https://doi.org/10.3390/ijms17010017
Perdicchia D, Christodoulou MS, Fumagalli G, Calogero F, Marucci C, Passarella D. Enzymatic Kinetic Resolution of 2-Piperidineethanol for the Enantioselective Targeted and Diversity Oriented Synthesis. International Journal of Molecular Sciences. 2016; 17(1):17. https://doi.org/10.3390/ijms17010017
Chicago/Turabian StylePerdicchia, Dario, Michael S. Christodoulou, Gaia Fumagalli, Francesco Calogero, Cristina Marucci, and Daniele Passarella. 2016. "Enzymatic Kinetic Resolution of 2-Piperidineethanol for the Enantioselective Targeted and Diversity Oriented Synthesis" International Journal of Molecular Sciences 17, no. 1: 17. https://doi.org/10.3390/ijms17010017