
Role for the Unfolded Protein Response in Heart Disease and Cardiac Arrhythmias

Abstract
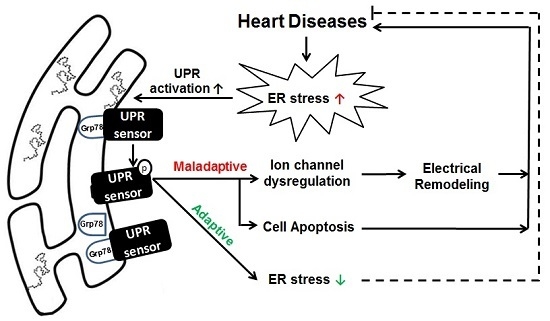
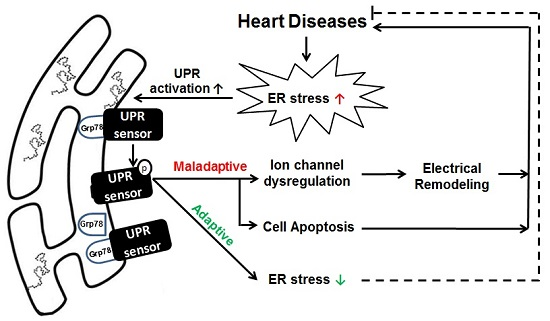
:
{kind=link}
{kind=link}
1. Introduction

2. The UPR in Heart Disease
2.1. UPR in Cardiac Hypertrophy
2.2. UPR in Ischemia
2.3. UPR in Heart Failure
2.4. UPR in Other Heart Diseases
3. UPR and Arrhythmias
4. Targeting the UPR in Heart Disease
4.1. Potential Benefits of UPR Inhibition
4.2. Potential Methods to Inhibit the UPR
5. Precautions on Targeting the UPR
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ma, Y.; Hendershot, L.M. ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 2004, 28, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Reimold, A.; Iwakoshi, N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.; Friend, D.; Grusby, M.; Alt, F.; Glimcher, L. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the α subunit of eukaryotic initiation factor 2 is required for activation of NF-kB in response to diverse cellular stresses. Mol. Cell. Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef] [PubMed]
- Byrd, A.; Aragon, I.; Brewer, J. MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J. Cell Biol. 2012, 196, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Xie, A.; Zhang, J.; Herman, A.M.; Jeong, E.M.; Gu, L.; Liu, M.; Yang, K.C.; Kamp, T.J.; Dudley, S.C. Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ. Arrhythm. Electrophysiol. 2013, 6, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Yoshii, S.; Hottori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Duffy, A.; O’Mahoney, M.E.; Logue, S.E.; Mylotte, L.A.; O’Brien, T.; Samali, A. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem. Biophys. Res. Commun. 2006, 349, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.F.; Yang, X.J. Endoplasmic reticulum stress response in spontaneously hypertensive rats is affected by myocardial ischemia reperfusion injury. Exp. Ther. Med. 2015, 9, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Avery, J.; Etzion, S.; DeBosch, B.J.; Jin, X.; Lupu, T.S.; Beitinjaneh, B.; Grand, J.; Kovacs, A.; Sambandam, N.; Muslin, A.J. TRB3 function in cardiac endoplasmic reticulum stress. Circ. Res. 2010, 106, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Doroudgar, S.; Thuerauf, D.J.; Marcinko, M.C.; Belmont, P.J.; Glembotski, C.C. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem. 2009, 284, 29735–29745. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.J.; Chen, W.J.; San Pedro, M.N.; Thuerauf, D.J.; Gellings Lowe, N.; Gude, N.; Hilton, B.; Wolkowicz, R.; Sussman, M.A.; Glembotski, C.C. Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene Derlin-3 in the ischemic heart. Circ. Res. 2010, 106, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Marcinko, M.; Gude, N.; Rubio, M.; Sussman, M.A.; Glembotski, C.C. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res. 2006, 99, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Minamino, T.; Fu, H.Y.; Asai, M.; Okuda, K.; Isomura, T.; Yamazaki, S.; Asano, Y.; Okada, K.; Tsukamoto, O.; et al. X-box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE-like element in cardiomyocytes. J. Mol. Cell. Cardiol. 2010, 48, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. BBA Mol. Cell. Biol. Lipids 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Sanbe, A.; Osinska, H.; Saffitz, J.E.; Glabe, C.G.; Kayed, R.; Maloyan, A.; Robbins, J. Desmin-related cardiomyopathy in transgenic mice: A cardiac amyloidosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10132–10136. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, Q.; Xu, W.; Cai, L. Endoplasmic reticulum stress and diabetic cardiomyopathy. Exp. Diabetes Res. 2012, 2012, 827971. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Patterson, C. Proteotoxicity and cardiac dysfunction—Alzheimer’s disease of the heart? N. Engl. J. Med. 2013, 368, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.Y.; Okada, K.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Sawada, T.; Okuda, K.; Asano, Y.; Sanada, S.; et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, O.; Higuchi, Y.; Hirotani, S.; Kashiwase, K.; Nakayama, H.; Hikoso, S.; Takeda, T.; Watanabe, T.; Asahi, M.; Taniike, M.; et al. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc. Natl. Acad. Sci. USA 2003, 100, 15883–15888. [Google Scholar] [CrossRef] [PubMed]
- Glembotski, C.C. Finding the missing link between the unfolded protein response and O-GlcNAcylation in the heart. Circ. Res. 2014, 115, 546–548. [Google Scholar] [CrossRef] [PubMed]
- Nickson, P.; Toth, A.; Erhardt, P. PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress. Cardiovasc. Res. 2007, 73, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.J.; Fernandez, R.; Thuerauf, D.; Whittaker, R.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 2006, 98, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Isodono, K.; Takahashi, T.; Imoto, H.; Nakanishi, N.; Ogata, T.; Asada, S.; Adachi, A.; Ueyama, T.; Oh, H.; Matsubara, H. PARM-1 is an endoplasmic reticulum molecue involved in endoplasmic reticulum stress-induced apoptosis in rat cardiac myocytes. PLoS ONE 2010, 5, e9746. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.; Roselló-Lletí, E.; Tarazón, E.; Molina-Navarro, M.M.; Martínez-Dolz, L.; González-Juanatey, J.; Lago, F.; Montoro-Mateos, J.D.; Salvador, A.; Rivera, M.; et al. Endoplasmic reticulum stress induces different molecular structural alterations in human dilated and ischemic cardiomyopathy. PLoS ONE 2014, 9, e107635. [Google Scholar] [CrossRef] [PubMed]
- Dally, S.; Monceau, V.; Corvazier, E.; Bredoux, R.; Raies, A.; Bobe, R.; del Monte, F.; Enouf, J. Compartmentalized expression of three novel sarco/endoplasmic reticulum Ca2+ATPase 3 isoforms including the switch to ER stress, SERCA3f, in non-failing and failing human heart. Cell Calcium 2009, 45, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kwak, D.; Lu, Z.; Xu, X.; Fassett, J.; Wang, H.; Wei, Y.; Cavener, D.R.; Hu, X.; Hall, J.; et al. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension 2014, 64, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, D.; Ren, J.; Yang, J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim. Biophys. Acta 2015, 1852, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, T.; Dai, H.; Liu, G.; Wang, H.; Sun, Y.; Zhang, Y.; Ge, Z. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J. Clin. Biochem. Nutr. 2007, 41, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, G.; Wang, Y.; Liu, Q.; Xu, W.; Tan, Y.; Cai, L. Diabetes- and angiotensin II-induced cardiac endoplasmic reticulum stress and cell death: Metallothionein protection. J. Cell. Mol. Med. 2009, 13, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Gilbert, S.A.B.; Li, Q.; Ren, J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. J. Mol. Cell. Cardiol. 2009, 47, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Kerkela, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Fukuoka, S.; Iwai, C.; Liu, J.; Sharma, V.K.; Sheu, S.S.; Fu, M.; Liang, C.S. Cardiomyocyte apoptosis in autoimmune cardiomyopathy: Mediated via endoplasmic reticulum stress and exaggerated by norepinephrine. Am. J. Physiol.-Heart. C 2007, 293, H1636–H1645. [Google Scholar] [CrossRef] [PubMed]
- Ceylan-Isik, A.F.; Sreejayan, N.; Ren, J. Endoplasmic reticulum chaperon tauroursodeoxycholic acid alleviates obesity-induced myocardial contractile dysfunction. J. Mol. Cell. Cardiol. 2011, 50, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Melkani, G.C.; Trujillo, A.S.; Ramos, R.; Bodmer, R.; Bernstein, S.I.; Ocorr, K. Huntington’s disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart. PLoS Genet. 2013, 9, e1004024. [Google Scholar] [CrossRef] [PubMed]
- Pattison, J.S.; Robbins, J. Protein misfolding and cardiac disease: Establishing cause and effect. Autophagy 2008, 4, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Pattison, J.S.; Sanbe, A.; Maloyan, A.; Osinska, H.; Klevitsky, R.; Robbins, J. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation 2008, 117, 2743–2751. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, S.F.; Picarote, G.; Fleming, J.V.; Carmo-Fonseca, M.; Azevedo, J.E.; de Sousa, M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J. Biol. Chem. 2007, 282, 27905–27912. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Takahashi, H.; Adachi, N.; Abe, N.; Shimahara, T.; Saito, N.; Sakai, N. Aggregate formation of mutant protein kinase C gamma found in spinocerebellar ataxia type 14 impairs ubiquitin-proteasome system and induces endoplasmic reticulum stress. Eur. J. Neurosci. 2007, 26, 3126–3140. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gu, L.; Sulkin, M.S.; Liu, H.; Jeong, E.M.; Greener, I.; Xie, A.; Efimov, I.R.; Dudley, S.C., Jr. Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J. Mol. Cell. Cardiol. 2013, 54, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Forrester, M.; Eyler, C.; Rich, J. Bacterial flavohemoglobin: A molecular tool to probe mammalian nitric oxide biology. BioTechniques 2011, 50, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Janse, M.J. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc. Res. 2004, 61, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Mercier, A.; Clément, R.; Harnois, T.; Bourmeyster, N.; Bois, P.; Chatelier, A. Nav1.5 channels can reach the plasma membrane through distinct N-glycosylation states. BBA-Gen. Subj. 2015, 1850, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.C.; Tanak, L.Y.; Wosniak, J., Jr.; Laurindo, F.R.M. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.; Nabeebaccus, A.; Shah, A.; Camargo, L.; Filho, S.; Lopes, L. Endoplasmic reticulum stress and nox-mediated reactive oxygen species signaling in the peripheral vasculature: Potential role in hypertension. Antioxid. Redox Signal. 2014, 20, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Xie, A.; Huang, S.C.; Zhou, A.; Zhang, J.; Herman, A.M.; Ghassemzadeh, S.; Jeong, E.M.; Kasturirangan, S.; Raicu, M.; et al. Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure/clinical perspective. Circulation 2011, 124, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.X.; Lin, L.; Ren, A.J.; Pan, X.J.; Chen, H.; Tang, C.S.; Yuan, W.J. HSP70 and GRP78 Induced by endothelin-1 pretreatment enhance tolerance to hypoxia in cultured neonatal rat cardiomyocytes. J. Cardiovasc. Pharmacol. 2004, 44, S117–S120. [Google Scholar] [CrossRef] [PubMed]
- Vitadello, M.; Penzo, D.; Petronilli, V.; Michieli, G.; Gomirato, S.; Menabò, R.; Di Lisa, F.; Gorza, L. Overexpression of the stress-protein Grp94 reduces cardiomyocyte necrosis due to calcium overload and simulated ischemia. FASEB J. 2003, 17, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.S.; Ayaub, E.A.; Zhou, W.; Yum, V.; Dickhout, J.G.; Ask, K. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int. J. Biochem. Cell Biol. 2015, 61, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The unexpected uses of urso- and tauroursodeoxycholic acid in the treatment of non-liver diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Kassan, M.; Galán, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Kim, J.K.; Chen, B.; Abdel-Latif, A.; Kitakaze, M.; Yan, L. Attenuation of ER stress prevents post-infarction-induced cardiac rupture and remodeling by modulating both cardiac apoptosis and fibrosis. Chem. Biol. Interact. 2015, 225, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nakajima, S.; Kato, H.; Gu, L.; Yoshitomi, T.; Nagai, K.; Shinmori, H.; Kokubo, S.; Kitamura, M. Selective, potent blockade of the IRE1 and ATF6 pathways by 4-phenylbutyric acid analogues. Br. J. Pharmacol. 2013, 170, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C. Alzheimer’s treatment breakthrough: British scientists pave way for simple pill to cure disease. Available online: http://www.independent.co.uk/news/uk/home-news/alzheimer-s-treatment-breakthrough-british-scientists-pave-way-for-simple-pill-to-cure-disease-8869716.html (accessed on 30 December 2015).
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.C.S.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I.; Kitakaze, M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ. Res. 2010, 107, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Zhu, W.; Li, Y.; Xin, P.; Li, J.; Liu, M.; Li, J.; Redington, A.N.; Wei, M. Apelin-13 protects the heart against ischemia-reperfusion injury through inhibition of ER-dependent apoptotic pathways in a time-dependent fashion. Am. J. Physiol.-Heart C 2011, 301, H1471–H1486. [Google Scholar] [CrossRef] [PubMed]
- Cornejo, V.; Pihán, P.; Vidal, R.; Hetz, C. Role of the unfolded protein response in organ physiology: Lessons from mouse models. IUBMB Life 2013, 65, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Yoshida, M.; Noguchi, S. Targeted disruption of CRE-binding factor TREB5 gene leads to cellular necrosis in cardiac myocytes at the embryonic stage. Biochem. Biophys. Res. Commun. 1999, 261, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The PERK eukaryotic initiation factor 2α kinase is required for the development of the skeletal systme, postnatal growth and the function and viability ofthe pancreas. Mol. Cell. Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6α-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell. 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Yamaguchi, S.; Tanji, Y.; Tominaga, R.; Ishigaki, Y.; Fukumoto, M.; Katagiri, H.; Mori, K.; Oka, Y.; Ishihara, H. Atf6α-null mice are glucose intolerant due to pancreatic β-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism 2012, 61, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Dudley, S.C., Jr. Role for the Unfolded Protein Response in Heart Disease and Cardiac Arrhythmias. Int. J. Mol. Sci. 2016, 17, 52. https://doi.org/10.3390/ijms17010052
Liu M, Dudley SC Jr. Role for the Unfolded Protein Response in Heart Disease and Cardiac Arrhythmias. International Journal of Molecular Sciences. 2016; 17(1):52. https://doi.org/10.3390/ijms17010052
Chicago/Turabian StyleLiu, Man, and Samuel C. Dudley, Jr. 2016. "Role for the Unfolded Protein Response in Heart Disease and Cardiac Arrhythmias" International Journal of Molecular Sciences 17, no. 1: 52. https://doi.org/10.3390/ijms17010052