
The Roles of RNase-L in Antimicrobial Immunity and the Cytoskeleton-Associated Innate Response

Abstract
:
1. Introduction
2. Activation of the 2′-5′-Linked Oligoadenylates (2-5A)/RNase-L Pathway

3. Regulation of RNase-L and Its Nuclease Activity
4. The Antimicrobial Actions of RNase-L
4.1. RNase-L Antiviral Activity
4.2. RNase-L-Mediated Antibacterial Activity
4.3. Alternative Cellular and Innate Immune Activities
5. RNase-L and the Cytoskeleton




6. Cytoskeletal Interactions as a Component of the Innate Immune Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Innate Immune Protein | Cytoskeleton Protein | Characterization of Interaction | Reference |
|---|---|---|---|
| Cytoskeleton regulated activation | |||
| NOD2 | Rac1 |
| Legrand-Poels et al. [136] |
| RIG-I | Unknown |
| Mukherjee et al. [137] |
| MAVS | FAK |
| Bozym et al. [139] |
| TBK1 | GEF-H1 |
| Chiang et al. [140] |
| Pyrin | Unknown |
| Kim et al. [135] |
| Caspase-1 | Flightless |
| Li et al. [141] |
| Innate regulation of the cytoskeleton | |||
| ASC | Dock2 |
| Ippagunta et al. [142] |
| RNase-L | Filamin A |
| Malathi et al. [28] |
| PKR | Gelsolin |
| Irving et al. [143] |
| Caspase-11 | Aip1 |
| Li et al. [144] |
| ADAP2 | Arf6 |
| Shu et al. [145] |
| Undetermined function of the interaction | |||
| IFITM1 | Occludin, Claudin1, and ZO-1 |
| Wilkins et al. [146] |
| RNase-L | IQGAP1 |
| Sato et al. [120] |
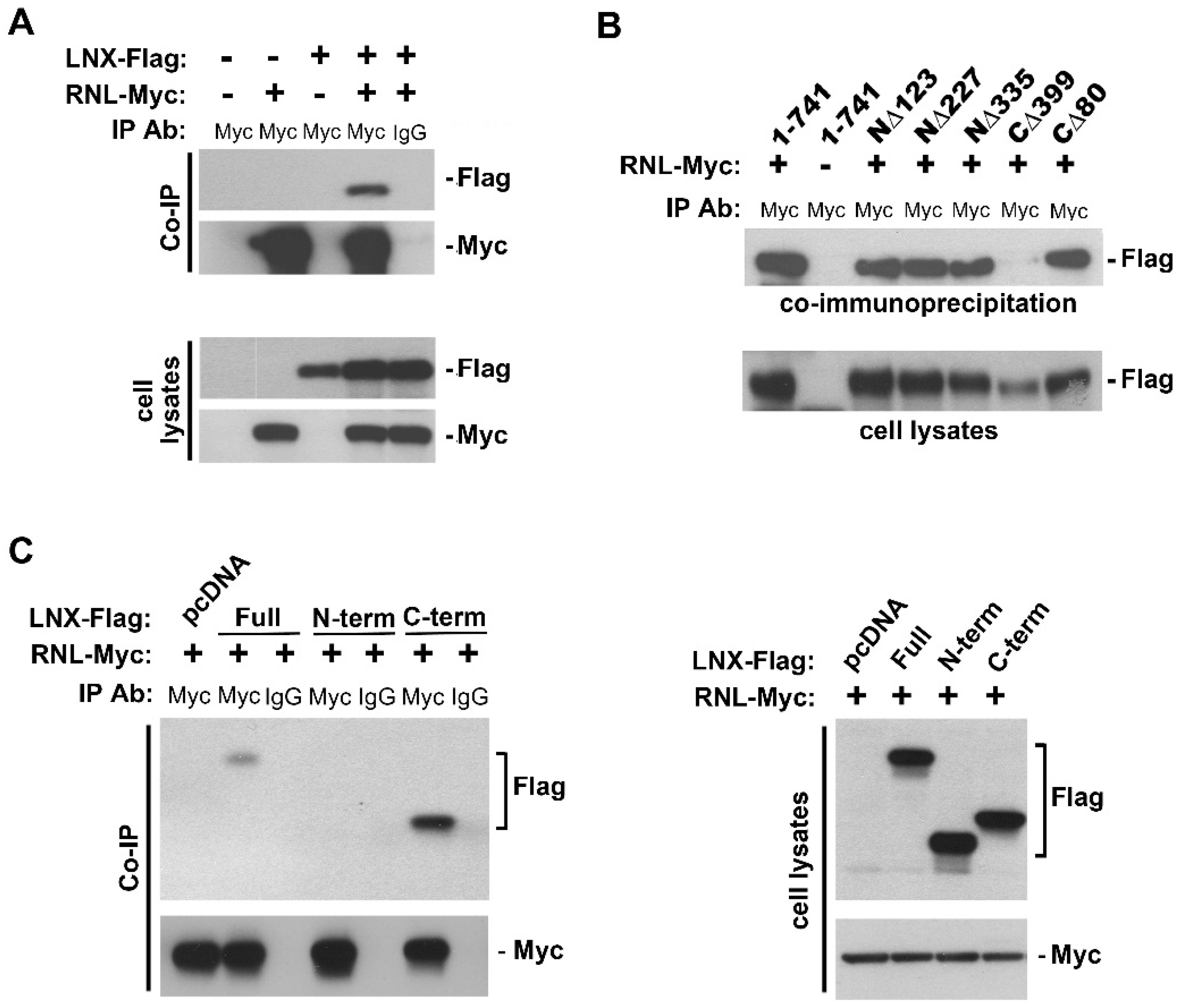
| LNX1 |
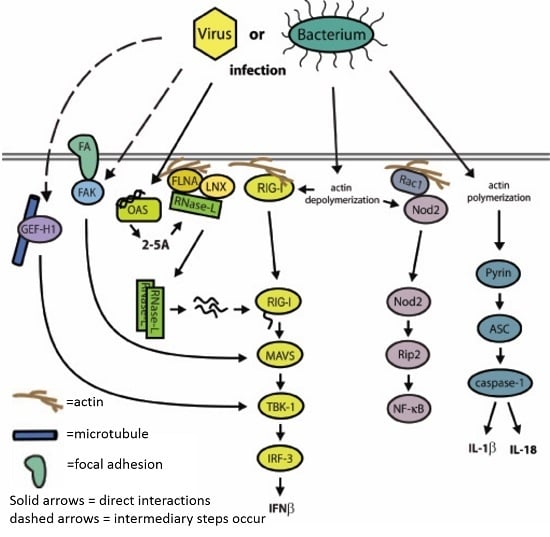
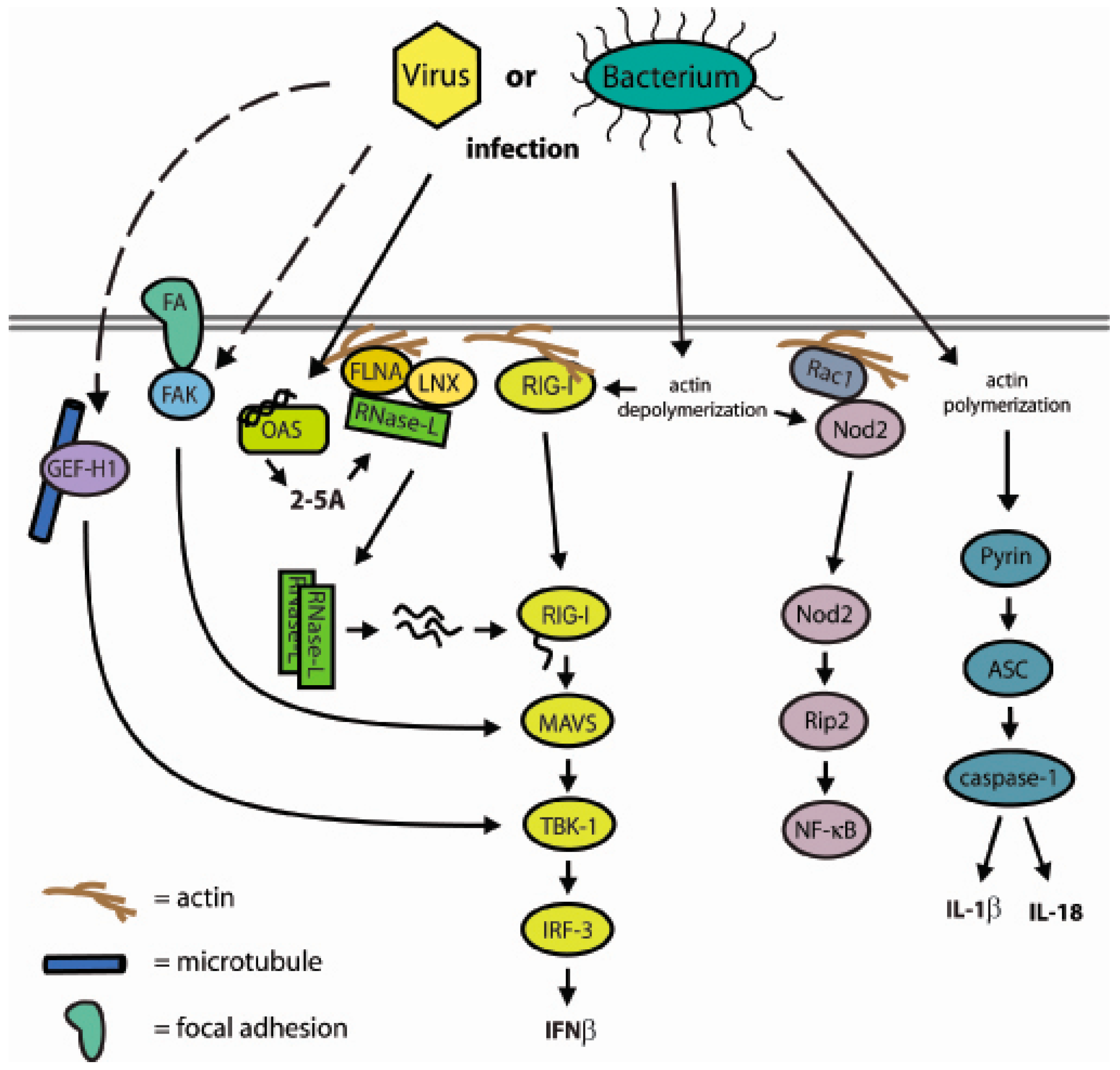
| Figure 2 and Figure 3 | |
7. A Network of Innate Immune Mediators Link Pathogen Sensing to Host Response via Cytoskeletal Associations: Emerging Model and Outstanding Questions
Acknowledgments
Conflicts of Interest
References
- Brand, S.; Zitzmann, K.; Dambacher, J.; Beigel, F.; Olszak, T.; Vlotides, G.; Eichhorst, S.T.; Goke, B.; Diepolder, H.; Auernhammer, C.J. SOCS-1 inhibits expression of the antiviral proteins 2′,5′-OAS and MxA induced by the novel interferon-λs IL-28a and IL-29. Biochem. Biophys. Res. Commun. 2005, 331, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon α,β, or γ using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G. On the discovery of interferon-inducible, double-stranded RNA activated enzymes: The 2′-5′oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev. 2007, 18, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Nakanishi, M.; Kusakabe, Y.; Goto, Y.; Kitade, Y.; Nakamura, K.T. Structural basis for recognition of 2′,5′-linked oligoadenylates by human ribonuclease L. EMBO J. 2004, 23, 3929–3938. [Google Scholar] [CrossRef] [PubMed]
- Baglioni, C.; Minks, M.A.; Maroney, P.A. Interferon action may be mediated by activation of a nuclease by pppa2′p5′A2′p5′A. Nature 1978, 273, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.J.; Williams, B.R. Inhibition of cell-free protein synthesis by pppa2′p5′A2′p5′A: A novel oligonucleotide synthesized by interferon-treated l cell extracts. Cell 1978, 13, 565–572. [Google Scholar] [CrossRef]
- Eppstein, D.A.; Samuel, C.E. Mechanism of interferon action. Properties of an interferon-mediated ribonucleolytic activity from mouse l929 cells. Virology 1978, 89, 240–251. [Google Scholar] [CrossRef]
- Hovanessian, A.G.; Brown, R.E.; Kerr, I.M. Synthesis of low molecular weight inhibitor of protein synthesis with enzyme from interferon-treated cells. Nature 1977, 268, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Ratner, L.; Wiegand, R.C.; Farrell, P.J.; Sen, G.C.; Cabrer, B.; Lengyel, P. Interferon, double-stranded RNA and RNA degradation. Fractionation of the endonucleaseint system into two macromolecular components; role of a small molecule in nuclease activation. Biochem. Biophys. Res. Commun. 1978, 81, 947–954. [Google Scholar] [CrossRef]
- Zhou, A.; Hassel, B.A.; Silverman, R.H. Expression cloning of 2-5A-dependent RNaase: A uniquely regulated mediator of interferon action. Cell 1993, 72, 753–765. [Google Scholar] [CrossRef]
- Carpten, J.; Nupponen, N.; Isaacs, S.; Sood, R.; Robbins, C.; Xu, J.; Faruque, M.; Moses, T.; Ewing, C.; Gillanders, E.; et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat. Genet. 2002, 30, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Castelli, J.C.; Hassel, B.A.; Maran, A.; Paranjape, J.; Hewitt, J.A.; Li, X.L.; Hsu, Y.T.; Silverman, R.H.; Youle, R.J. The role of 2′-5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998, 5, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Castelli, J.C.; Hassel, B.A.; Wood, K.A.; Li, X.L.; Amemiya, K.; Dalakas, M.C.; Torrence, P.F.; Youle, R.J. A study of the interferon antiviral mechanism: Apoptosis activation by the 2-5A system. J. Exp. Med. 1997, 186, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Hassel, B.A.; Zhou, A.; Sotomayor, C.; Maran, A.; Silverman, R.H. A dominant negative mutant of 2-5A-dependent RNase suppresses antiproliferative and antiviral effects of interferon. EMBO J. 1993, 12, 3297–3304. [Google Scholar] [PubMed]
- Zhou, A.; Paranjape, J.; Brown, T.L.; Nie, H.; Naik, S.; Dong, B.; Chang, A.; Trapp, B.; Fairchild, R.; Colmenares, C.; et al. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997, 16, 6355–6363. [Google Scholar] [CrossRef] [PubMed]
- Demettre, E.; Bastide, L.; D’Haese, A.; De Smet, K.; De Meirleir, K.; Tiev, K.P.; Englebienne, P.; Lebleu, B. Ribonuclease L proteolysis in peripheral blood mononuclear cells of chronic fatigue syndrome patients. J. Biol. Chem. 2002, 277, 35746–35751. [Google Scholar] [CrossRef] [PubMed]
- Shetzline, S.E.; Suhadolnik, R.J. Characterization of a 2′,5′-oligoadenylate (2-5A)-dependent 37-kDa RNase L: Azido photoaffinity labeling and 2-5A-dependent activation. J. Biol. Chem. 2001, 276, 23707–23711. [Google Scholar] [CrossRef] [PubMed]
- Suhadolnik, R.J.; Peterson, D.L.; O’Brien, K.; Cheney, P.R.; Herst, C.V.; Reichenbach, N.L.; Kon, N.; Horvath, S.E.; Iacono, K.T.; Adelson, M.E.; et al. Biochemical evidence for a novel low molecular weight 2-5A-dependent RNase L in chronic fatigue syndrome. J. Interferon Cytokine Res. 1997, 17, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Li, X.L.; Judge, C.S.; Zhou, A.; Jha, B.K.; Shelby, S.; Zhou, L.; Silverman, R.H.; Hassel, B.A. Role of 2-5A-dependent RNase-L in senescence and longevity. Oncogene 2006, 26, 3081–3088. [Google Scholar] [CrossRef] [PubMed]
- Fabre, O.; Breuker, C.; Amouzou, C.; Salehzada, T.; Kitzmann, M.; Mercier, J.; Bisbal, C. Defects in TLR3 expression and RNase L activation lead to decreased mnsod expression and insulin resistance in muscle cells of obese people. Cell Death Dis. 2014, 5, e1136. [Google Scholar] [CrossRef] [PubMed]
- Fabre, O.; Salehzada, T.; Lambert, K.; Boo Seok, Y.; Zhou, A.; Mercier, J.; Bisbal, C. RNase L controls terminal adipocyte differentiation, lipids storage and insulin sensitivity via CHOP10 mRNA regulation. Cell Death Differ. 2012, 19, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Ireland, D.D.; Stohlman, S.A.; Hinton, D.R.; Kapil, P.; Silverman, R.H.; Atkinson, R.A.; Bergmann, C.C. RNase L mediated protection from virus induced demyelination. PLoS Pathog. 2009, 5, e1000602. [Google Scholar] [CrossRef] [PubMed]
- Long, T.M.; Chakrabarti, A.; Ezelle, H.J.; Brennan-Laun, S.E.; Raufman, J.P.; Polyakova, I.; Silverman, R.H.; Hassel, B.A. RNase-L deficiency exacerbates experimental colitis and colitis-associated cancer. Inflamm. Bowel Dis. 2013, 19, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Yi, X.; Zipris, D.; Liu, H.; Zhang, L.; Zheng, Q.; Malathi, K.; Jin, G.; Zhou, A. RNase L contributes to experimentally induced type 1 diabetes onset in mice. J. Endocrinol. 2014, 223, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Laun, S.E.; Ezelle, H.J.; Li, X.L.; Hassel, B.A. RNase-L control of cellular mRNAs: Roles in biologic functions and mechanisms of substrate targeting. J. Interferon Cytokine Res. 2014, 34, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Ezelle, H.J.; Kang, T.J.; Zhang, L.; Shirey, K.A.; Harro, J.; Hasday, J.D.; Mohapatra, S.K.; Crasta, O.R.; Vogel, S.N.; et al. An essential role for the antiviral endoribonuclease, RNase-L, in antibacterial immunity. Proc. Natl. Acad. Sci. USA 2008, 105, 20816–20821. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Siddiqui, M.A.; Dayal, S.; Naji, M.; Ezelle, H.J.; Zeng, C.; Zhou, A.; Hassel, B.A. RNase L interacts with filamin a to regulate actin dynamics and barrier function for viral entry. MBio 2014, 5, e02012. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Malathi, K. RNase L induces autophagy via c-jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J. Biol. Chem. 2012, 287, 43651–43664. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Mukherjee, S.; Manivannan, P.; Malathi, K. RNase L cleavage products promote switch from autophagy to apoptosis by caspase-mediated cleavage of beclin-1. Int J. Mol. Sci. 2015, 16, 17611–17636. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H.; Zhou, A.; Auerbach, M.B.; Kish, D.; Gorbachev, A.; Fairchild, R.L. Skin allograft rejection is suppressed in mice lacking the antiviral enzyme, 2′,5′-oligoadenylate-dependent RNase L. Viral Immunol. 2002, 15, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Zeng, C.; Liu, H.; Chen, X.; Zhang, P.; Yun, B.S.; Jin, G.; Zhou, A. Lack of RNase L attenuates macrophage functions. PLoS ONE 2013, 8, e81269. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Rebouillat, D.; Hovanessian, A.G. The human 2′,5′-oligoadenylate synthetase family: Interferon-induced proteins with unique enzymatic properties. J. Interferon Cytokine Res. 1999, 19, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G.; Justesen, J. The human 2′-5′oligoadenylate synthetase family: Unique interferon-inducible enzymes catalyzing 2′-5′ instead of 3′-5′ phosphodiester bond formation. Biochimie 2007, 89, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.K.; et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, R.; Norby, P.L.; Martensen, P.M.; Jorgensen, P.; James, M.C.; Jacobsen, C.; Moestrup, S.K.; Clemens, M.J.; Justesen, J. Activation of 2′-5′ oligoadenylate synthetase by single-stranded and double-stranded RNA aptamers. J. Biol. Chem. 1998, 273, 3236–3246. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, R.J.; Jha, B.K.; Malathi, K.; Varambally, S.; Chinnaiyan, A.M.; Silverman, R.H. Selection and cloning of poly(rC)-binding protein 2 and raf kinase inhibitor protein RNA activators of 2′,5′-oligoadenylate synthetase from prostate cancer cells. Nucleic Acids Res. 2006, 34, 6684–6695. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, H.R.; Sheetz, P.C.; Iogal, S.S.; Brodsky, I.; Kariko, K.; Li, S.W.; Suhadolnik, R.J.; Sobol, R.W. Heterogeneous nuclear RNA from hairy cell leukemia patients activates 2′,5′-oligoadenylate synthetase. Anticancer Res. 1991, 11, 1927–1932. [Google Scholar] [PubMed]
- Andersen, J.B.; Mazan-Mamczarz, K.; Zhan, M.; Gorospe, M.; Hassel, B.A. Ribosomal protein mRNAs are primary targets of regulation in RNase-L-induced senescence. RNA Biol. 2009, 6, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Madsen, B.E.; Ramos, E.M.; Boulard, M.; Duda, K.; Overgaard, J.; Nordsmark, M.; Wiuf, C.; Hansen, L.L. Germline mutation in RNase L predicts increased risk of head and neck, uterine cervix and breast cancer. PLoS ONE 2008, 3, e2492. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Silverman, R.H. A bipartite model of 2-5A-dependent RNase L. J. Biol. Chem. 1997, 272, 22236–22242. [Google Scholar] [CrossRef] [PubMed]
- Knight, M.; Cayley, P.J.; Silverman, R.H.; Wreschner, D.H.; Gilbert, C.S.; Brown, R.E.; Kerr, I.M. Radioimmune, radiobinding and hplc analysis of 2-5A and related oligonucleotides from intact cells. Nature 1980, 288, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Nakahara, K.; Ohtsuka, T.; Yoshida, S.; Kawaguchi, J.; Fujita, Y.; Ozeki, Y.; Hara, A.; Yoshimura, C.; Furukawa, H.; et al. Identification of 2′-phosphodiesterase, which plays a role in the 2-5A system regulated by interferon. J. Biol. Chem. 2004, 279, 37832–37841. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H.; Wreschner, D.H.; Gilbert, C.S.; Kerr, I.M. Synthesis, characterization and properties of ppp(A2′p)nApCp and related high-specific-activity 32P-labelled derivatives of ppp(A2′p)nA. Eur. J. Biochem. 1981, 115, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Bledsoe, R.; Chai, J.; Daka, P.; Deng, H.; Ding, Y.; Harris-Gurley, S.; Kryn, L.H.; Nartey, E.; Nichols, J.; et al. The role of phosphodiesterase 12 (PDE12) as a negative regulator of the innate immune response and the discovery of antiviral inhibitors. J. Biol. Chem. 2015, 290, 19681–19696. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Donovan, J.; Rath, S.; Whitney, G.; Chitrakar, A.; Korennykh, A. Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 2014, 343, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Whitney, G.; Donovan, J.; Korennykh, A. Innate immune messenger 2-5A tethers human RNase L into active high-order complexes. Cell Rep. 2012, 2, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zeqiraj, E.; Dong, B.; Jha, B.K.; Duffy, N.M.; Orlicky, S.; Thevakumaran, N.; Talukdar, M.; Pillon, M.C.; Ceccarelli, D.F.; et al. Dimeric structure of pseudokinase RNase L bound to 2-5A reveals a basis for interferon-induced antiviral activity. Mol. Cell 2014, 53, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Floyd-Smith, G.; Slattery, E.; Lengyel, P. Interferon action: RNA cleavage pattern of a (2′-5′)oligoadenylate--dependent endonuclease. Science 1981, 212, 1030–1032. [Google Scholar] [CrossRef] [PubMed]
- Wreschner, D.H.; McCauley, J.W.; Skehel, J.J.; Kerr, I.M. Interferon action—Sequence specificity of the ppp(A2′p)nA-dependent ribonuclease. Nature 1981, 289, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Blackford, J.A.; Hassel, B.A. RNase L mediates the antiviral effect of interferon through a selective reduction in viral RNA during encephalomyocarditis virus infection. J. Virol. 1998, 72, 2752–2759. [Google Scholar] [PubMed]
- Brennan-Laun, S.E.; Li, X.L.; Ezelle, H.J.; Venkataraman, T.; Blackshear, P.J.; Wilson, G.M.; Hassel, B.A. RNase L attenuates mitogen-stimulated gene expression via transcriptional and post-transcriptional mechanisms to limit the proliferative response. J. Biol. Chem. 2014, 289, 33629–33643. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Paranjape, J.M.; Bulanova, E.; Shim, M.; Guenther-Johnson, J.M.; Faber, P.W.; Eling, T.E.; Williams, B.R.; Silverman, R.H. A transcriptional signaling pathway in the IFN system mediated by 2′-5′-oligoadenylate activation of RNase L. Proc. Natl. Acad. Sci. USA 2005, 102, 14533–14538. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Molinaro, R.J.; Malathi, K.; Silverman, R.H. Mapping of the human RNase L promoter and expression in cancer and normal cells. J. Interferon Cytokine Res. 2005, 25, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Maurin, V.; Brinza, L.; Baguet, J.; Plantamura, E.; Schicklin, S.; Chambion, S.; Macari, C.; Tomkowiak, M.; Deniaud, E.; Leverrier, Y.; et al. Overexpression of the transcription factor sp1 activates the OAS-RNase L-RIG-I pathway. PLoS ONE 2015, 10, e0118551. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Ezelle, H.J.; Venkataraman, T.; Lapidus, R.G.; Scheibner, K.A.; Hassel, B.A. Regulation of human RNase-L by the mir-29 family reveals a novel oncogenic role in chronic myelogenous leukemia. J. Interferon Cytokine Res. 2013, 33, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Andersen, J.B.; Ezelle, H.J.; Wilson, G.M.; Hassel, B.A. Post-transcriptional regulation of RNase-L expression is mediated by the 3′-untranslated region of its mRNA. J. Biol. Chem. 2007, 282, 7950–7960. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Gorospe, M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev. RNA 2010, 1, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Chase, B.I.; Zhou, Y.; Xiang, Y.; Silverman, R.H.; Zhou, A. Proteasome-mediated degradation of RNase L in response to phorbol-12-myristate-13-acetate (PMA) treatment of mouse l929 cells. J. Interferon Cytokine Res. 2003, 23, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, U.; Takahashi, S.; Ishizu, A.; Miyatake, Y.; Gohda, A.; Suzuki, S.; Ono, A.; Ohara, J.; Baba, T.; Murata, S.; et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 2012, 180, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Webb, J.D.; Kramer, H.B.; Kessler, B.M.; Ratcliffe, P.J. Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell. Proteom. 2009, 8, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Bisbal, C.; Martinand, C.; Silhol, M.; Lebleu, B.; Salehzada, T. Cloning and characterization of a RNase L inhibitor. A new component of the interferon-regulated 2-5A pathway. J. Biol. Chem. 1995, 270, 13308–13317. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Lai, R.; Nielsen, K.; Fekete, C.A.; Qiu, H.; Hinnebusch, A.G. The essential ATP-binding cassette protein RLI1 functions in translation by promoting preinitiation complex assembly. J. Biol. Chem. 2004, 279, 42157–42168. [Google Scholar] [CrossRef] [PubMed]
- Khoshnevis, S.; Gross, T.; Rotte, C.; Baierlein, C.; Ficner, R.; Krebber, H. The iron-sulphur protein RNase Linhibitor functions in translation termination. EMBO Rep. 2010, 11, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Kispal, G.; Sipos, K.; Lange, H.; Fekete, Z.; Bedekovics, T.; Janaky, T.; Bassler, J.; Aguilar Netz, D.J.; Balk, J.; Rotte, C.; et al. Biogenesis of cytosolic ribosomes requires the essential iron-sulphur protein RLI1P and mitochondria. EMBO J. 2005, 24, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, F.; Bisbal, C.; Silhol, M.; Martinand, C.; Lebleu, B.; Salehzada, T. The 2-5A/RNase L/RNase L inhibitor (RLI) [correction of (RNI)] pathway regulates mitochondrial mRNAs stability in interferon α-treated h9 cells. J. Biol. Chem. 2001, 276, 48473–48482. [Google Scholar] [PubMed]
- Malathi, K.; Paranjape, J.M.; Ganapathi, R.; Silverman, R.H. HPC1/RNase L mediates apoptosis of prostate cancer cells treated with 2′,5′-oligoadenylates, topoisomerase i inhibitors, and tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2004, 64, 9144–9151. [Google Scholar] [CrossRef] [PubMed]
- Martinand, C.; Montavon, C.; Salehzada, T.; Silhol, M.; Lebleu, B.; Bisbal, C. RNase L inhibitor is induced during human immunodeficiency virus type 1 infection and down regulates the 2-5A/RNase L pathway in human T cells. J. Virol. 1999, 73, 290–296. [Google Scholar] [PubMed]
- Martinand, C.; Salehzada, T.; Silhol, M.; Lebleu, B.; Bisbal, C. RNase L inhibitor (RLI) antisense constructions block partially the down regulation of the 2-5A/RNase L pathway in encephalomyocarditis-virus-(EMCV)-infected cells. Eur. J. Biochem. 1998, 254, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, F.; Salehzada, T.; Bisbal, C.; Dougherty, J.P.; Peltz, S.W. A newly discovered function for RNase L in regulating translation termination. Nat. Struct. Mol. Biol. 2005, 12, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Salehzada, T.; Silhol, M.; Steff, A.M.; Lebleu, B.; Bisbal, C. 2′,5′-oligoadenylate-dependent RNase L is a dimer of regulatory and catalytic subunits. J. Biol. Chem. 1993, 268, 7733–7740. [Google Scholar] [PubMed]
- Inge-Vechtomov, S.; Zhouravleva, G.; Philippe, M. Eukaryotic release factors (ERFS) history. Biol. Cell 2003, 95, 195–209. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Mehrabian, Z.; Li, X.L.; Hassel, B. RNase Lregulates the stability of mitochondrial DNA-encoded mRNAs in mouse embryo fibroblasts. Biochem. Biophys. Res. Commun. 2004, 325, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, F.; Silhol, M.; Salehzada, T.; Bisbal, C. Regulation of mitochondrial mRNA stability by RNase L is translation-dependent and controls IFNα-induced apoptosis. Cell Death Differ. 2007, 14, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Besse, S.; Rebouillat, D.; Marie, I.; Puvion-Dutilleul, F.; Hovanessian, A.G. Ultrastructural localization of interferon-inducible double-stranded RNA-activated enzymes in human cells. Exp. Cell Res. 1998, 239, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, J.B.; Andersen, K.R.; Kjaer, K.H.; Durand, F.; Faou, P.; Vestergaard, A.L.; Talbo, G.H.; Hoogenraad, N.; Brodersen, D.E.; Justesen, J.; et al. Human 2′-phosphodiesterase localizes to the mitochondrial matrix with a putative function in mitochondrial RNA turnover. Nucleic Acids Res. 2011, 39, 3754–3770. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.E.; Kuwano, Y.; Alkharouf, N.; Blackshear, P.J.; Gorospe, M.; Wilson, G.M. The mRNA-destabilizing protein tristetraprolin is suppressed in many cancers, altering tumorigenic phenotypes and patient prognosis. Cancer Res. 2009, 69, 5168–5176. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A.; Blackshear, P.J. Tristetraprolin (TTP): Interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim. Biophys. Acta 2013, 1829, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Komano, J.; Saitoh, Y.; Yamaoka, S.; Kozaki, T.; Misawa, T.; Takahama, M.; Satoh, T.; Takeuchi, O.; Yamamoto, N.; et al. Zinc-finger antiviral protein mediates retinoic acid inducible gene I-like receptor-independent antiviral response to murine leukemia virus. Proc. Natl. Acad. Sci. USA 2013, 110, 12379–12384. [Google Scholar] [CrossRef] [PubMed]
- Uehata, T.; Akira, S. mRNA degradation by the endoribonuclease Regnase-1/ZC3H12a/MCPIP-1. Biochim. Biophys. Acta 2013, 1829, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Banerjee, S.; Franchi, L.; Loo, Y.M.; Gale, M., Jr.; Nunez, G.; Silverman, R.H. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 2015, 17, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Long, T.M.; Nisa, S.; Donnenberg, M.S.; Hassel, B.A. Enteropathogenic escherichia coli inhibits type I interferon- and RNase L-mediated host defense to disrupt intestinal epithelial cell barrier function. Infect. Immun. 2014, 82, 2802–2814. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [PubMed]
- Gribaudo, G.; Lembo, D.; Cavallo, G.; Landolfo, S.; Lengyel, P. Interferon action: Binding of viral RNA to the 40-kilodalton 2′-5′-oligoadenylate synthetase in interferon-treated hela cells infected with encephalomyocarditis virus. J. Virol. 1991, 65, 1748–1757. [Google Scholar] [PubMed]
- Williams, B.R.; Golgher, R.R.; Brown, R.E.; Gilbert, C.S.; Kerr, I.M. Natural occurrence of 2-5A in interferon-treated emc virus-infected l cells. Nature 1979, 282, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Q.; Townsend, H.L.; Jha, B.K.; Paranjape, J.M.; Silverman, R.H.; Barton, D.J. A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J. Virol. 2007, 81, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Sorgeloos, F.; Jha, B.K.; Silverman, R.H.; Michiels, T. Evasion of antiviral innate immunity by theiler’s virus l* protein through direct inhibition of RNase L. PLoS Pathog. 2013, 9, e1003474. [Google Scholar] [CrossRef] [PubMed]
- Townsend, H.L.; Jha, B.K.; Han, J.Q.; Maluf, N.K.; Silverman, R.H.; Barton, D.J. A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA 2008, 14, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Townsend, H.L.; Jha, B.K.; Silverman, R.H.; Barton, D.J. A putative loop E motif and an H-H kissing loop interaction are conserved and functional features in a group C enterovirus RNA that inhibits ribonuclease L. RNA Biol. 2008, 5, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Q.; Barton, D.J. Activation and evasion of the antiviral 2′-5′ oligoadenylate synthetase/ribonuclease L pathway by hepatitis c virus mRNA. RNA 2002, 8, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Washenberger, C.L.; Han, J.Q.; Kechris, K.J.; Jha, B.K.; Silverman, R.H.; Barton, D.J. Hepatitis c virus RNA: Dinucleotide frequencies and cleavage by RNase L. Virus Res. 2007, 130, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Mihm, U.; Ackermann, O.; Welsch, C.; Herrmann, E.; Hofmann, W.P.; Grigorian, N.; Welker, M.W.; Lengauer, T.; Zeuzem, S.; Sarrazin, C. Clinical relevance of the 2′-5′-oligoadenylate synthetase/RNase L system for treatment response in chronic hepatitis c. J. Hepatol. 2009, 50, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.A.; Banerjee, S.; Chakrabarti, A.; Garcia-Sastre, A.; Hesselberth, J.R.; Silverman, R.H.; Barton, D.J. RNase L targets distinct sites in influenza a virus RNAs. J. Virol. 2015, 89, 2764–2776. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef] [PubMed]
- Ezelle, H.J.; Hassel, B.A.; Torrence, P.F. Mechanisms of IFN resistance by influenza virus. In Combating the Threat of Pandemic Influenza: Drug Discovery Approaches; Torrence, P.F., Ed.; Wiley-Interscience: Hoboken, NJ, USA, 2007; pp. 73–97. [Google Scholar]
- Wu, J.M.; Chiao, J.W.; Maayan, S. Diagnostic value of the determination of an interferon-induced enzyme activity: Decreased 2′,5′-oligoadenylate dependent binding protein activity in aids patient lymphocytes. AIDS Res. 1986, 2, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Jha, B.K.; Wu, A.; Elliott, R.; Ziebuhr, J.; Gorbalenya, A.E.; Silverman, R.H.; Weiss, S.R. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 2012, 11, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Rusch, L.; Zhou, A.; Silverman, R.H. Caspase-dependent apoptosis by 2′,5′-oligoadenylate activation of RNase L is enhanced by IFN-β. J. Interferon Cytokine Res. 2000, 20, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Ghosh, P.K.; Banerjee, S.; Gaughan, C.; Silverman, R.H. RNase L triggers autophagy in response to viral infections. J. Virol. 2012, 86, 11311–11321. [Google Scholar] [CrossRef] [PubMed]
- Gordy, C.; He, Y.W. The crosstalk between autophagy and apoptosis: Where does this lead? Protein Cell 2012, 3, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.P.; Marshall, H.D.; Laidlaw, B.J.; Staron, M.M.; Kaech, S.M.; Craft, J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity 2014, 40, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.L.; Wilson, T.J.; Strauch, P.; Colonna, M.; Pelanda, R.; Torres, R.M. Type I IFN enhances follicular b cell contribution to the T cell-independent antibody response. J. Exp. Med. 2010, 207, 1485–1500. [Google Scholar] [CrossRef] [PubMed]
- Claes, A.K.; Zhou, J.Y.; Philpott, D.J. NOD-like receptors: Guardians of intestinal mucosal barriers. Physiology 2015, 30, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Dugan, J.W.; Albor, A.; David, L.; Fowlkes, J.; Blackledge, M.T.; Martin, T.M.; Planck, S.R.; Rosenzweig, H.L.; Rosenbaum, J.T.; Davey, M.P. Nucleotide oligomerization domain-2 interacts with 2′-5′-oligoadenylate synthetase type 2 and enhances RNase-L function in thp-1 cells. Mol. Immunol. 2009, 47, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Bayard, B.A.; Gabrion, J.B. 2′,5′-oligoadenylate-dependent RNase located in nuclei: Biochemical characterization and subcellular distribution of the nuclease in human and murine cells. Biochem. J. 1993, 296, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Rath, P.C. Expression of mRNA and protein-protein interaction of the antiviral endoribonuclease RNase L in mouse spleen. Int. J. Biol. Macromol. 2014, 69, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Tnani, M.; Aliau, S.; Bayard, B. Localization of a molecular form of interferon-regulated RNase L in the cytoskeleton. J. Interferon Cytokine Res. 1998, 18, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Cohen, L.A.; Donaldson, J.G. Arf1 and arf6 promote ventral actin structures formed by acute activation of protein kinase c and src. Cytoskeleton 2014, 71, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Sumagin, R.; Robin, A.Z.; Nusrat, A.; Parkos, C.A. Activation of PKCβII by PMA facilitates enhanced epithelial wound repair through increased cell spreading and migration. PLoS ONE 2013, 8, e55775. [Google Scholar] [CrossRef] [PubMed]
- Tanos, B.E.; Perez Bay, A.E.; Salvarezza, S.; Vivanco, I.; Mellinghoff, I.; Osman, M.; Sacks, D.B.; Rodriguez-Boulan, E. IQGAP1 controls tight junction formation through differential regulation of claudin recruitment. J. Cell Sci. 2015, 128, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Abel, A.M.; Schuldt, K.M.; Rajasekaran, K.; Hwang, D.; Riese, M.J.; Rao, S.; Thakar, M.S.; Malarkannan, S. IQGAP1: Insights into the function of a molecular puppeteer. Mol. Immunol. 2015, 65, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Ezelle, H.J.; Hassel, B.A. Pathologic effects of RNase-L dysregulation in immunity and proliferative control. Front. Biosci. 2012, 4, 767–786. [Google Scholar] [CrossRef]
- Kim, H.; White, C.D.; Sacks, D.B. IQGAP1 in microbial pathogenesis: Targeting the actin cytoskeleton. FEBS Lett. 2011, 585, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Yokogawa, T.; Takatori, S.; Goda, K.; Hiramoto, A.; Sato, A.; Kitade, Y.; Sasaki, T.; Matsuda, A.; Fukushima, M.; et al. Role of RNase L in apoptosis induced by 1-(3-C-ethynyl-β-d-ribo-pentofuranosyl)cytosine. Cancer Chemother. Pharmacol. 2009, 63, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Naito, T.; Hiramoto, A.; Goda, K.; Omi, T.; Kitade, Y.; Sasaki, T.; Matsuda, A.; Fukushima, M.; Wataya, Y.; et al. Association of RNase L with a ras GTPase-activating-like protein IQGAP1 in mediating the apoptosis of a human cancer cell-line. FEBS J. 2010, 277, 4464–4473. [Google Scholar] [CrossRef] [PubMed]
- Dho, S.E.; Jacob, S.; Wolting, C.D.; French, M.B.; Rohrschneider, L.R.; McGlade, C.J. The mammalian numb phosphotyrosine-binding domain. Characterization of binding specificity and identification of a novel pdz domain-containing numb binding protein, lnx. J. Biol. Chem. 1998, 273, 9179–9187. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; McGill, M.A.; Dermer, M.; Dho, S.E.; Wolting, C.D.; McGlade, C.J. Lnx functions as a ring type e3 ubiquitin ligase that targets the cell fate determinant numb for ubiquitin-dependent degradation. EMBO J. 2002, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Song, E.; Ma, S.; Wang, X.; Gao, S.; Shao, C.; Hu, S.; Jia, L.; Tian, R.; Xu, T.; et al. Proteomics strategy to identify substrates of lnx, a pdz domain-containing e3 ubiquitin ligase. J. Proteom. Res. 2012, 11, 4847–4862. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, J.; Zhao, W.; Hu, G.; Cheng, H.; Kang, Y.; Xie, Y.; Lu, Y. Characterization of human lnx, a novel Ligand of Numb protein X that is downregulated in human gliomas. Int. J. Biochem. Cell Biol. 2005, 37, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Wu, Q.; Liu, W.; Zhu, F.; Qiu, F.; Zhou, Q.; Fan, J.; Dong, X.; Yu, X. Ectopic expression of Ligand-of-Numb protein X promoted TGF-β induced epithelial to mesenchymal transition of proximal tubular epithelial cells. Biochim. Biophys. Acta 2009, 1792, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Iwamoto, N.; Sasaki, H.; Ohashi, M.; Oda, Y.; Tsukita, S.; Furuse, M. The e3 ubiquitin ligase lnx1p80 promotes the removal of claudins from tight junctions in mdck cells. J. Cell Sci. 2009, 122, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Baumgartner, M.; Radziwill, G.; Dennler, J.; Moelling, K. C-src is a pdz interaction partner and substrate of the e3 ubiquitin ligase Ligand-of-Numb protein X1. FEBS Lett. 2007, 581, 5131–5136. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Gu, S.; Li, Y.; Ji, C.; Xie, Y.; Mao, Y. A global genomic view on lnx siRNA-mediated cell cycle arrest. Mol. Biol. Rep. 2011, 38, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Sun, Y.; Gu, S.; Ji, C.; Zhao, W.; Xie, Y.; Mao, Y. LNX (Ligand of Numb-Protein X) interacts with RhoC, both of which regulate AP-1-mediated transcriptional activation. Mol. Biol. Rep. 2010, 37, 2431–2437. [Google Scholar] [CrossRef] [PubMed]
- Kansaku, A.; Hirabayashi, S.; Mori, H.; Fujiwara, N.; Kawata, A.; Ikeda, M.; Rokukawa, C.; Kurihara, H.; Hata, Y. Ligand-of-Numb protein X is an endocytic scaffold for junctional adhesion molecule 4. Oncogene 2006, 25, 5071–5084. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.; Raschperger, E.; Philipson, L.; Pettersson, R.F.; Sollerbrant, K. The cell surface protein coxsackie- and adenovirus receptor (CAR) directly associates with the Ligand-of-Numb protein-X2 (LNX2). Exp. Cell Res. 2005, 309, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Sollerbrant, K.; Raschperger, E.; Mirza, M.; Engstrom, U.; Philipson, L.; Ljungdahl, P.O.; Pettersson, R.F. The coxsackievirus and adenovirus receptor (CAR) forms a complex with the pdz domain-containing protein Ligand-of-Numb protein-X (LNX). J. Biol. Chem. 2003, 278, 7439–7444. [Google Scholar] [CrossRef] [PubMed]
- Haglund, C.M.; Welch, M.D. Pathogens and polymers: Microbe-host interactions illuminate the cytoskeleton. J. Cell Biol. 2011, 195, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.; Wu, Y. Viral exploitation of actin: Force-generation and scaffolding functions in viral infection. Virol. Sin. 2014, 29, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.L.; Chae, J.J.; Park, Y.H.; De Nardo, D.; Stirzaker, R.A.; Ko, H.J.; Tye, H.; Cengia, L.; DiRago, L.; Metcalf, D.; et al. Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1β. J. Exp. Med. 2015, 212, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Legrand-Poels, S.; Kustermans, G.; Bex, F.; Kremmer, E.; Kufer, T.A.; Piette, J. Modulation of NOD2-dependent NF-κB signaling by the actin cytoskeleton. J. Cell Sci. 2007, 120, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Shen, L.; Weber, C.R.; Turner, J.R.; Kim, K.S.; Wang, T.; Coyne, C.B. Retinoic acid-induced gene-1 (RIG-I) associates with the actin cytoskeleton via caspase activation and recruitment domain-dependent interactions. J. Biol. Chem. 2009, 284, 6486–6494. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Sun, L.; Zhang, H.; Liu, Q.; Liu, Y.; Qin, L.; Shi, G.; Hu, J.H.; Xu, A.; Sun, Y.P.; et al. An essential role for RIG-I in Toll-like receptor-stimulated phagocytosis. Cell Host Microbe 2009, 6, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Bozym, R.A.; Delorme-Axford, E.; Harris, K.; Morosky, S.; Ikizler, M.; Dermody, T.S.; Sarkar, S.N.; Coyne, C.B. Focal adhesion kinase is a component of antiviral RIG-I-like receptor signaling. Cell Host Microbe 2012, 11, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.S.; Zhao, Y.; Song, J.H.; Liu, S.; Wang, N.; Terhorst, C.; Sharpe, A.H.; Basavappa, M.; Jeffrey, K.L.; Reinecker, H.C. GEF-H1 controls microtubule-dependent sensing of nucleic acids for antiviral host defenses. Nat. Immunol. 2014, 15, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yin, H.L.; Yuan, J. Flightless-i regulates proinflammatory caspases by selectively modulating intracellular localization and caspase activity. J. Cell Biol. 2008, 181, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Ippagunta, S.K.; Malireddi, R.K.; Shaw, P.J.; Neale, G.A.; Vande Walle, L.; Green, D.R.; Fukui, Y.; Lamkanfi, M.; Kanneganti, T.D. The inflammasome adaptor ASC regulates the function of adaptive immune cells by controlling Dock2-mediated rac activation and actin polymerization. Nat. Immunol. 2011, 12, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Irving, A.T.; Wang, D.; Vasilevski, O.; Latchoumanin, O.; Kozer, N.; Clayton, A.H.; Szczepny, A.; Morimoto, H.; Xu, D.; Williams, B.R.; et al. Regulation of actin dynamics by protein kinase R control of gelsolin enforces basal innate immune defense. Immunity 2012, 36, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Brieher, W.M.; Scimone, M.L.; Kang, S.J.; Zhu, H.; Yin, H.; von Andrian, U.H.; Mitchison, T.; Yuan, J. Caspase-11 regulates cell migration by promoting Aip1-cofilin-mediated actin depolymerization. Nat. Cell Biol. 2007, 9, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Shu, Q.; Lennemann, N.J.; Sarkar, S.N.; Sadovsky, Y.; Coyne, C.B. ADAP2 is an interferon stimulated gene that restricts RNA virus entry. PLoS Pathog. 2015, 11, e1005150. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, C.; Woodward, J.; Lau, D.T.; Barnes, A.; Joyce, M.; McFarlane, N.; McKeating, J.A.; Tyrrell, D.L.; Gale, M., Jr. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 2013, 57, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, A.; Alonso, C.; Kurachi, K.; Gupta, S.; Lesser, C.F.; McCormick, B.A.; Reinecker, H.C. GEF-H1 mediated control of NOD1 dependent NF-κB activation by shigella effectors. PLoS Pathog. 2008, 4, e1000228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Alonso, C.; Ballester, I.; Song, J.H.; Chang, S.Y.; Guleng, B.; Arihiro, S.; Murray, P.J.; Xavier, R.; Kobayashi, K.S.; et al. Control of NOD2 and Rip2-dependent innate immune activation by GEF-H1. Inflamm. Bowel Dis. 2012, 18, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Coyne, C.B. Picornavirus entry. Adv. Exp. Med. Biol. 2013, 790, 24–41. [Google Scholar] [PubMed]
- Zhu, Y.Z.; Qian, X.J.; Zhao, P.; Qi, Z.T. How hepatitis c virus invades hepatocytes: The mystery of viral entry. World J. Gastroenterol. 2014, 20, 3457–3467. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.S.; Takahasi, K.; Ng, C.S.; Ouda, R.; Onomoto, K.; Yoneyama, M.; Lai, J.C.; Lattmann, S.; Nagamine, Y.; Matsui, T.; et al. DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog. 2014, 10, e1004012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, R.C.; O’Neill, L.A. New insights into the regulation of signalling by Toll-like receptors and NOD-like receptors. J. Innate Immun. 2010, 2, 406–421. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezelle, H.J.; Malathi, K.; Hassel, B.A. The Roles of RNase-L in Antimicrobial Immunity and the Cytoskeleton-Associated Innate Response. Int. J. Mol. Sci. 2016, 17, 74. https://doi.org/10.3390/ijms17010074
Ezelle HJ, Malathi K, Hassel BA. The Roles of RNase-L in Antimicrobial Immunity and the Cytoskeleton-Associated Innate Response. International Journal of Molecular Sciences. 2016; 17(1):74. https://doi.org/10.3390/ijms17010074
Chicago/Turabian StyleEzelle, Heather J., Krishnamurthy Malathi, and Bret A. Hassel. 2016. "The Roles of RNase-L in Antimicrobial Immunity and the Cytoskeleton-Associated Innate Response" International Journal of Molecular Sciences 17, no. 1: 74. https://doi.org/10.3390/ijms17010074