
The Importance of Thrombin in Cerebral Injury and Disease

Abstract
:
1. Introduction
2. Thrombin Signalling in Health and Disease
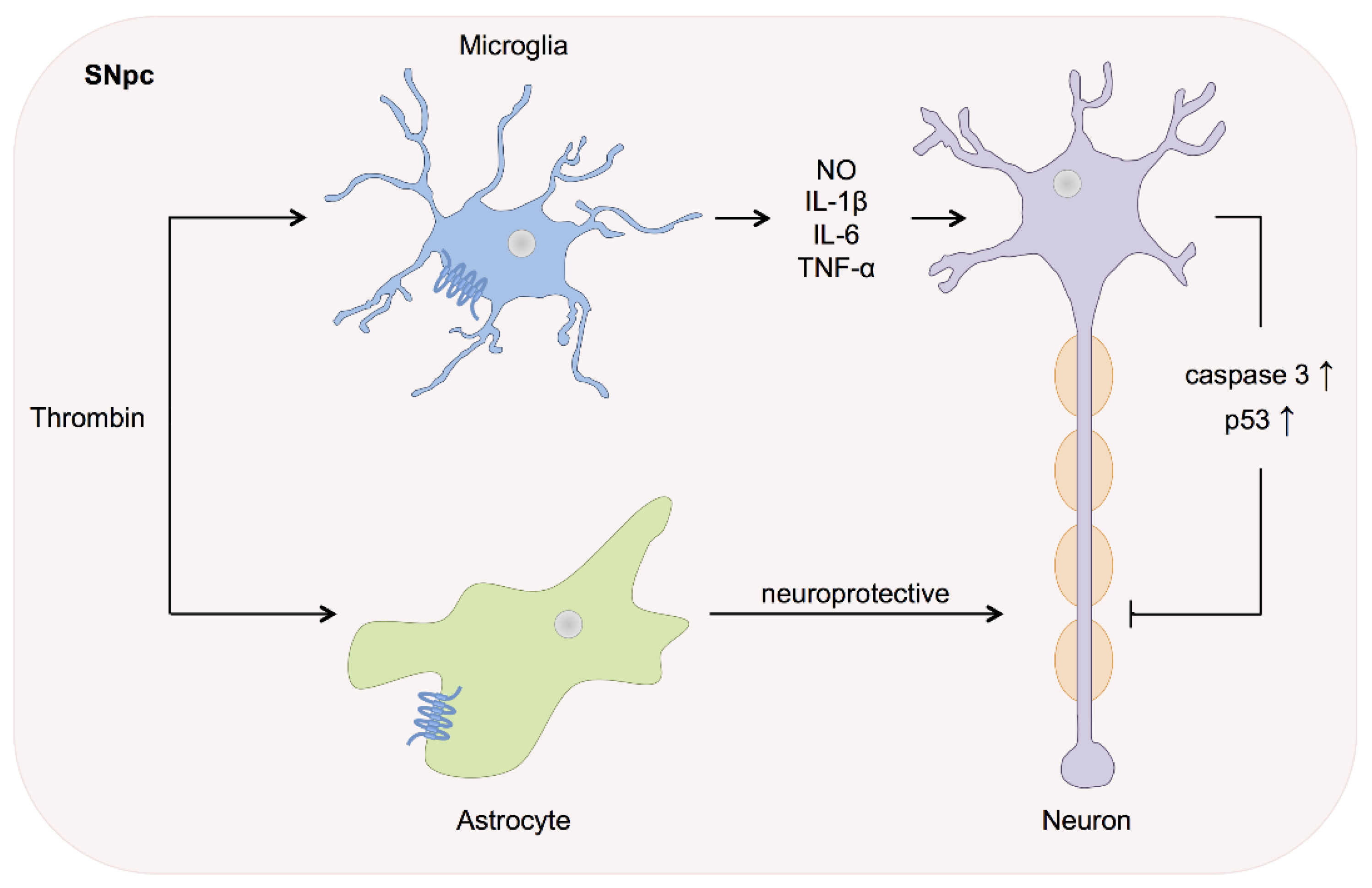
3. Thrombin in Neurodegenerative Disease



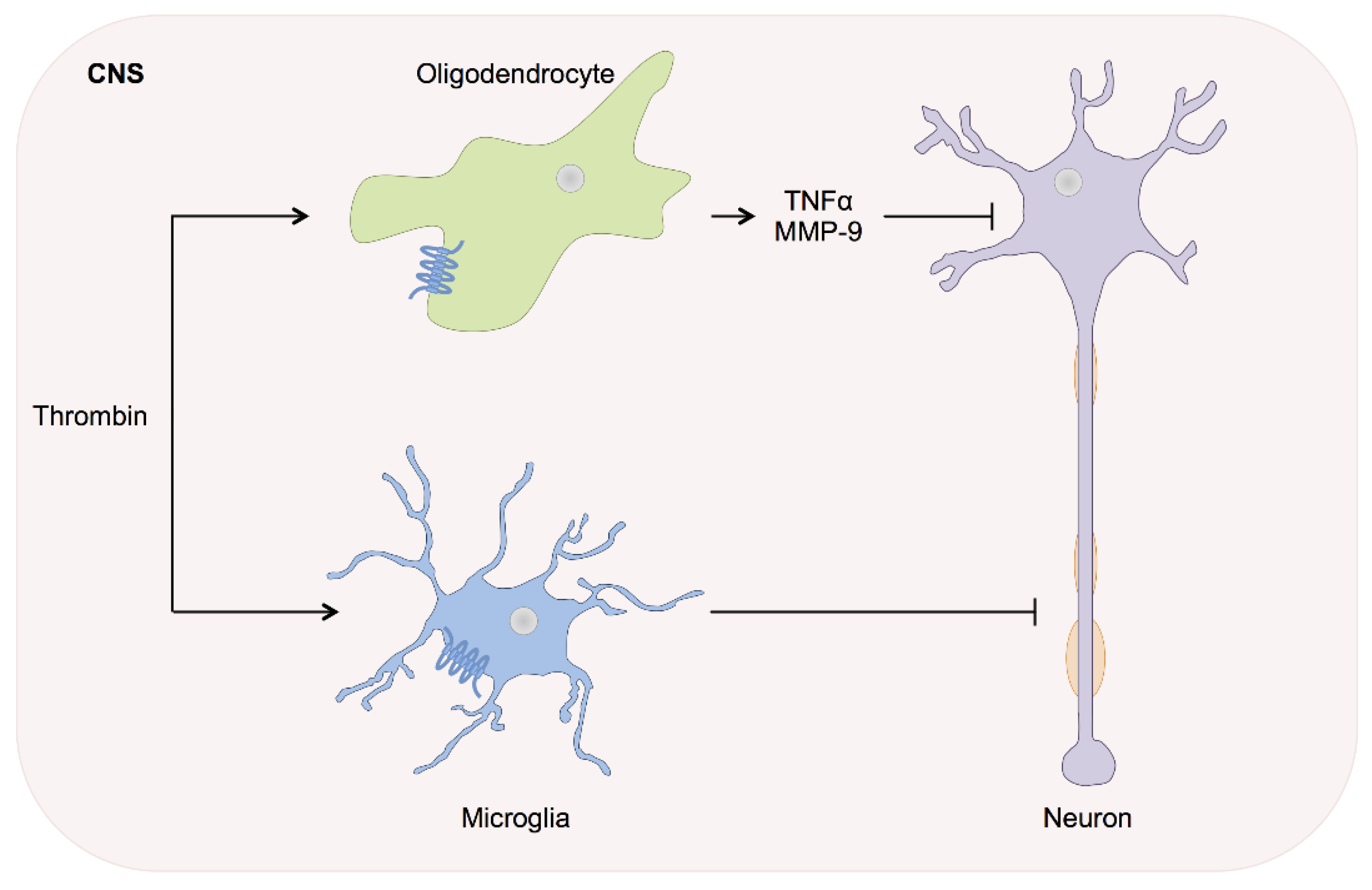
4. Thrombin in Stroke and Intracerebral Haemorrhage

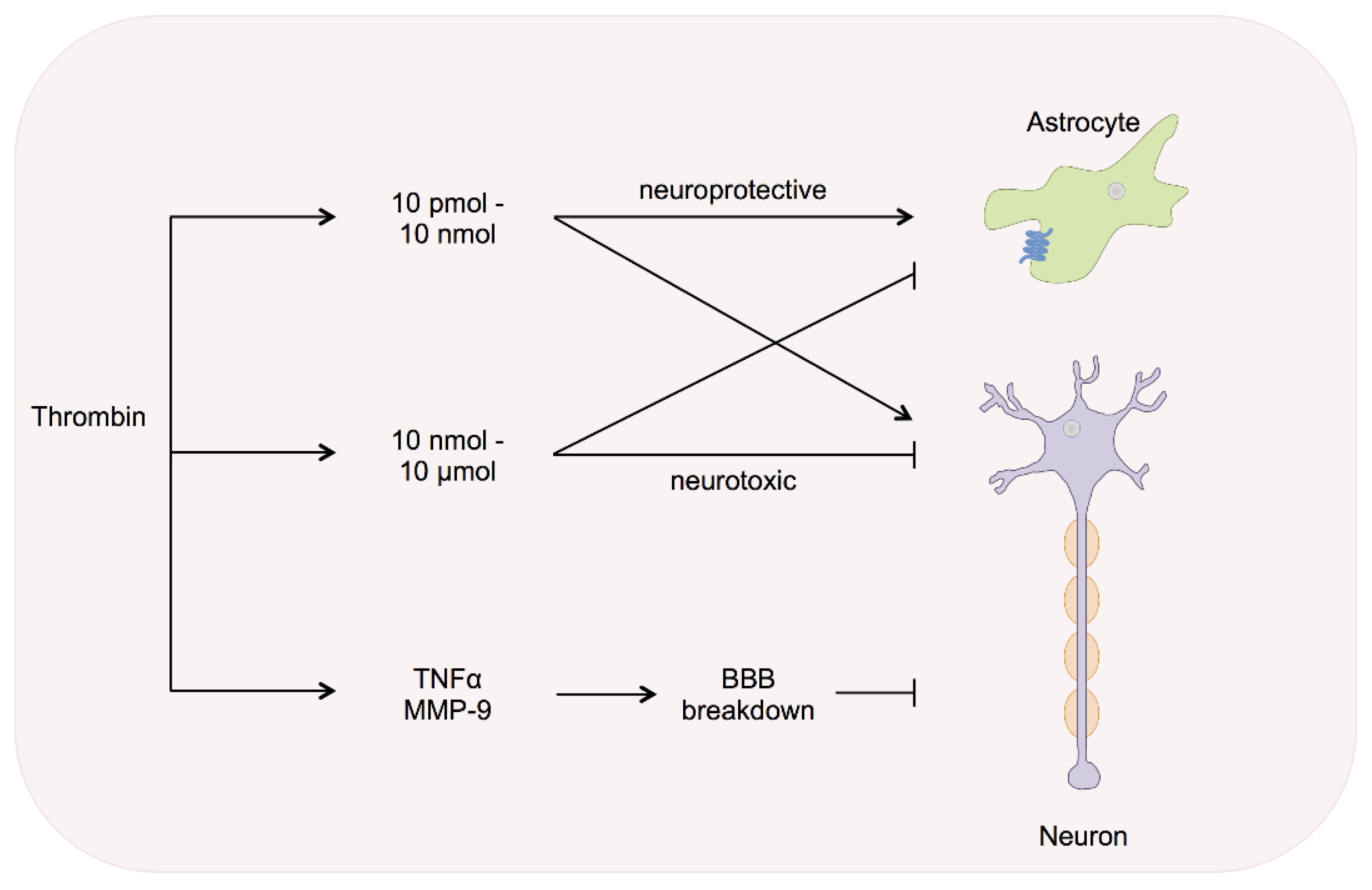
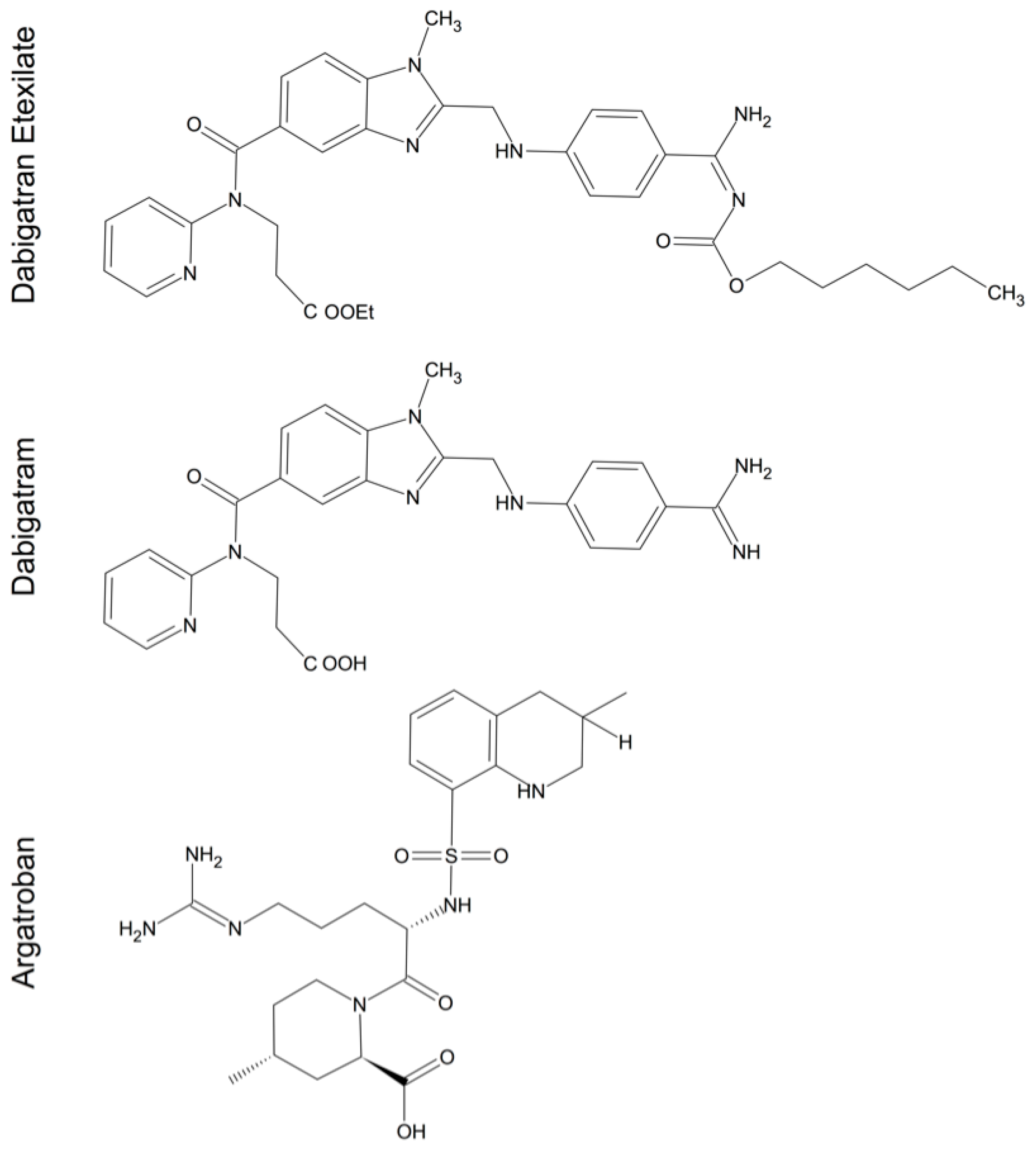
5. Thrombin Inhibitors in Cerebral Injury and Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| β-APP | amyloid-β precursor protein |
| Aβ | amyloid beta |
| AD | Alzheimer’s disease |
| AT | antithrombin |
| BBB | blood-brain-barrier |
| CDK 4 | Cyclin-dependent kinase 4 |
| CINC-1 | cytokine-induced neutrophil chemoattractant-1 |
| CNS | central nervous system |
| DTI | direct acting thrombin inhibitors |
| EAE | experimental autoimmune encephalitis |
| ERK1 | extracellular signal-regulated protein kinase 1 |
| F | factor |
| GFAP | glial fibrillary acidic protein |
| GP | glycoprotein |
| GRO | growth-regulated oncogene |
| HIF-1α | hypoxia inducing factor 1α |
| HIV | human immunodeficiency virus |
| HIVD | HIV dementia |
| ICH | intracerebral haemorrhage |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| JNK | N-terminal kinase |
| MAPK | mitogen- activated protein kinase |
| MMP-9 | matrix metalloproteinase-9 |
| MS | multiple sclerosis |
| NFTs | neurofibrillary tangles |
| NMDA | N-methyl-d-aspartate |
| NO | nitric oxid |
| OGD | glucose deprivation |
| 6-OHDA | 6-hydroxydopamine |
| PARs | protease-activated receptors |
| PD | Parkinson’s disease |
| PI 3-kinase | phosphatidylinositol 3-kinase |
| PKC | protein kinase C |
| PN-1 | protease nexin-1 |
| PTX | pertussis toxin |
| RAS | rat sarcoma protein |
| ROS | reactive oxygen species |
| SNpc | substantia nigra pars compacta |
| TNF-α | tumor necrosis factor-α |
| TPC | thrombin preconditioning |
| ZO | zonula occludens |
References
- Fenton, J.W., 2nd. Thrombin. Ann. N. Y. Acad. Sci. 1986, 485, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Grand, R.J.; Turnell, A.S.; Grabham, P.W. Cellular consequences of thrombin-receptor activation. Biochem. J. 1996, 313, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Strukova, S.M. Role of platelets and serine proteinases in coupling of blood coagulation and inflammation. Biochem. Biokhimiia 2004, 69, 1067–1081. [Google Scholar] [CrossRef]
- Ossovskaya, V.S.; Bunnett, N.W. Protease-activated receptors: Contribution to physiology and disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, M.; Buddenkotte, J.; Shpacovitch, V.; Rattenholl, A.; Moormann, C.; Vergnolle, N.; Luger, T.A.; Hollenberg, M.D. Proteinase-activated receptors: Transducers of proteinase-mediated signaling in inflammation and immune response. Endocr. Rev. 2005, 26, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Bock, P.E.; Olson, S.T.; Bjork, I. Inactivation of thrombin by antithrombin is accompanied by inactivation of regulatory exosite I. J. Biol. Chem. 1997, 272, 19837–19845. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.T.; Bode, W. The clot thickens: Clues provided by thrombin structure. Trends Biochem. Sci. 1995, 20, 23–28. [Google Scholar] [CrossRef]
- Lechtenberg, B.C.; Freund, S.M.; Huntington, J.A. GpIbalpha interacts exclusively with exosite II of thrombin. J. Mol. Biol. 2014, 426, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.P.; Royston, D. Thrombin generation and its inhibition: A review of the scientific basis and mechanism of action of anticoagulant therapies. Br. J. Anaesth. 2002, 88, 848–863. [Google Scholar] [CrossRef] [PubMed]
- Davie, E.W.; Fujikawa, K.; Kisiel, W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 1991, 30, 10363–10370. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, E.; Reiser, G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thromb. Haemost. 2008, 100, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Deschepper, C.F.; Bigornia, V.; Berens, M.E.; Lapointe, M.C. Production of thrombin and antithrombin III by brain and astroglial cell cultures. Brain Res. Mol. Brain Res. 1991, 11, 355–358. [Google Scholar] [CrossRef]
- Niclou, S.P.; Suidan, H.S.; Pavlik, A.; Vejsada, R.; Monard, D. Changes in the expression of protease-activated receptor 1 and protease nexin-1 mRNA during rat nervous system development and after nerve lesion. Eur. J. Neurosci. 1998, 10, 1590–1607. [Google Scholar] [CrossRef] [PubMed]
- Dihanich, M.; Kaser, M.; Reinhard, E.; Cunningham, D.; Monard, D. Prothrombin mRNA is expressed by cells of the nervous system. Neuron 1991, 6, 575–581. [Google Scholar] [CrossRef]
- Jamison, C.S.; Degen, S.J. Prenatal and postnatal expression of mRNA coding for rat prothrombin. Biochim. Biophys. Acta 1991, 1088, 208–216. [Google Scholar] [CrossRef]
- Yamada, T.; Nagai, Y. Immunohistochemical studies of human tissues with antibody to factor Xa. Histochem. J. 1996, 28, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Shikamoto, Y.; Morita, T. Expression of factor X in both the rat brain and cells of the central nervous system. FEBS Lett. 1999, 463, 387–389. [Google Scholar] [CrossRef]
- Wang, H.; Reiser, G. Thrombin signaling in the brain: The role of protease-activated receptors. Biol. Chem. 2003, 384, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.R.; Colon, G.P.; Betz, A.L.; Keep, R.F.; Kim, S.; Hoff, J.T. Edema from intracerebral hemorrhage: The role of thrombin. J. Neurosurg. 1996, 84, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.R.; Kawai, N.; Kim, S.; Sagher, O.; Hoff, J.T. Mechanisms of edema formation after intracerebral hemorrhage: Effects of thrombin on cerebral blood flow, blood-brain barrier permeability, and cell survival in a rat model. J. Neurosurg. 1997, 86, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Golde, T.; Kroon, S.N.; Perry, G. Serine protease inhibitor antithrombin III and its messenger RNA in the pathogenesis of Alzheimer’s disease. Am. J. Pathol. 1993, 143, 886–893. [Google Scholar] [PubMed]
- Walker, D.G.; Yasuhara, O.; Patston, P.A.; McGeer, E.G.; McGeer, P.L. Complement C1 inhibitor is produced by brain tissue and is cleaved in Alzheimer disease. Brain Res. 1995, 675, 75–82. [Google Scholar] [CrossRef]
- Mansuy, I.M.; van der Putten, H.; Schmid, P.; Meins, M.; Botteri, F.M.; Monard, D. Variable and multiple expression of Protease Nexin-1 during mouse organogenesis and nervous system development. Development 1993, 119, 1119–1134. [Google Scholar] [PubMed]
- Pindon, A.; Hantai, D.; Jandrot-Perrus, M.; Festoff, B.W. Novel expression and localization of active thrombomodulin on the surface of mouse brain astrocytes. Glia 1997, 19, 259–268. [Google Scholar] [CrossRef]
- Hua, Y.; Xi, G.; Keep, R.F.; Wu, J.; Jiang, Y.; Hoff, J.T. Plasminogen activator inhibitor-1 induction after experimental intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2002, 22, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Suzuki, M.; Kim, T.; Wagner, S.L.; Cunningham, D.D. Protease nexin-1. Localization in the human brain suggests a protective role against extravasated serine proteases. Am. J. Pathol. 1990, 137, 741–747. [Google Scholar] [PubMed]
- Arai, T.; Miklossy, J.; Klegeris, A.; Guo, J.P.; McGeer, P.L. Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 2006, 65, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Protease-activated receptors start a family. Proc. Natl. Acad. Sci. USA 1994, 91, 9200–9202. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Molecular mechanisms of thrombin signaling. Semin. Hematol. 1994, 31, 270–277. [Google Scholar] [PubMed]
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, H.; Connolly, A.J.; Zeng, D.; Kahn, M.L.; Zheng, Y.W.; Timmons, C.; Tram, T.; Coughlin, S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997, 386, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, S.; Emilsson, K.; Wahlestedt, C.; Sundelin, J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 9208–9212. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ubl, J.J.; Reiser, G. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia 2002, 37, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.R.; Seatter, M.J.; Kanke, T.; Hunter, G.D.; Plevin, R. Proteinase-activated receptors. Pharmacol. Rev. 2001, 53, 245–282. [Google Scholar] [PubMed]
- Striggow, F.; Riek-Burchardt, M.; Kiesel, A.; Schmidt, W.; Henrich-Noack, P.; Breder, J.; Krug, M.; Reymann, K.G.; Reiser, G. Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur. J. Neurosci. 2001, 14, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Ubl, J.J.; Vohringer, C.; Reiser, G. Co-existence of two types of [Ca2+]i-inducing protease-activated receptors (PAR-1 and PAR-2) in rat astrocytes and C6 glioma cells. Neuroscience 1998, 86, 597–609. [Google Scholar] [CrossRef]
- Weinstein, J.R.; Gold, S.J.; Cunningham, D.D.; Gall, C.M. Cellular localization of thrombin receptor mRNA in rat brain: Expression by mesencephalic dopaminergic neurons and codistribution with prothrombin mRNA. J. Neurosci. 1995, 15, 2906–2919. [Google Scholar] [PubMed]
- Wang, H.; Ubl, J.J.; Stricker, R.; Reiser, G. Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am. J. Physiol. Cell Physiol. 2002, 283, C1351–C1364. [Google Scholar] [CrossRef] [PubMed]
- Sinnreich, M.; Meins, M.; Niclou, S.P.; Suidan, H.S.; Monard, D. Prothrombin overexpressed in post-natal neurones requires blood factors for activation in the mouse brain. J. Neurochem. 2004, 88, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.D.; Gurwitz, D. Proteolytic regulation of neurite outgrowth from neuroblastoma cells by thrombin and protease nexin-1. J. Cell. Biochem. 1989, 39, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Grabham, P.; Cunningham, D.D. Thrombin receptor activation stimulates astrocyte proliferation and reversal of stellation by distinct pathways: Involvement of tyrosine phosphorylation. J. Neurochem. 1995, 64, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Pai, K.S.; Mahajan, V.B.; Lau, A.; Cunningham, D.D. Thrombin receptor signaling to cytoskeleton requires Hsp90. J. Biol. Chem. 2001, 276, 32642–32647. [Google Scholar] [CrossRef] [PubMed]
- Jalink, K.; van Corven, E.J.; Hengeveld, T.; Morii, N.; Narumiya, S.; Moolenaar, W.H. Inhibition of lysophosphatidate- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein Rho. J. Cell Biol. 1994, 126, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, A.; Reiser, G.; Reymann, K.G. Protease-activated receptor-1 induces generation of new microglia in the dentate gyrus of traumatised hippocampal slice cultures. Neurosci. Lett. 2007, 415, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Wu, M.; Ameenuddin, S.; Anderson, H.E.; Zoloty, J.E.; Citron, B.A.; Andrade-Gordon, P.; Festoff, B.W. Participation of protease-activated receptor-1 in thrombin-induced microglial activation. J. Neurochem. 2002, 80, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Debeir, T.; Gueugnon, J.; Vige, X.; Benavides, J. Transduction mechanisms involved in thrombin receptor-induced nerve growth factor secretion and cell division in primary cultures of astrocytes. J. Neurochem. 1996, 66, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Jalink, K.; Moolenaar, W.H. Thrombin receptor activation causes rapid neural cell rounding and neurite retraction independent of classic second messengers. J. Cell Biol. 1992, 118, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, H.; Costa, T.; Clouse, K.A.; Pluta, R.M.; Ogino, Y.; Coligan, J.E.; Burd, P.R. Thrombin is a regulator of astrocytic endothelin-1. Brain Res. 1993, 600, 201–207. [Google Scholar] [CrossRef]
- Vaughan, P.J.; Pike, C.J.; Cotman, C.W.; Cunningham, D.D. Thrombin receptor activation protects neurons and astrocytes from cell death produced by environmental insults. J. Neurosci. 1995, 15, 5389–5401. [Google Scholar] [PubMed]
- Gingrich, M.B.; Junge, C.E.; Lyuboslavsky, P.; Traynelis, S.F. Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 2000, 20, 4582–4595. [Google Scholar] [PubMed]
- Gingrich, M.B.; Traynelis, S.F. Serine proteases and brain damage—Is there a link? Trends Neurosci. 2000, 23, 399–407. [Google Scholar] [CrossRef]
- Davis-Salinas, J.; Saporito-Irwin, S.M.; Donovan, F.M.; Cunningham, D.D.; van Nostrand, W.E. Thrombin receptor activation induces secretion and nonamyloidogenic processing of amyloid β-protein precursor. J. Biol. Chem. 1994, 269, 22623–22627. [Google Scholar] [PubMed]
- Meli, R.; Raso, G.M.; Cicala, C.; Esposito, E.; Fiorino, F.; Cirino, G. Thrombin and PAR-1 activating peptide increase iNOS expression in cytokine-stimulated C6 glioma cells. J. Neurochem. 2001, 79, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Moller, T.; Hanisch, U.K.; Ransom, B.R. Thrombin-induced activation of cultured rodent microglia. J. Neurochem. 2000, 75, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Wu, M.; Citron, B.A.; Gao, C.; Festoff, B.W. Persistent protease-activated receptor 4 signaling mediates thrombin-induced microglial activation. J. Biol. Chem. 2003, 278, 31177–31183. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Joe, E.H.; Kim, S.U.; Jin, B.K. Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. J. Neurosci. 2003, 23, 5877–5886. [Google Scholar] [PubMed]
- Choi, S.H.; Lee, D.Y.; Chung, E.S.; Hong, Y.B.; Kim, S.U.; Jin, B.K. Inhibition of thrombin-induced microglial activation and NADPH oxidase by minocycline protects dopaminergic neurons in the substantia nigra in vivo. J. Neurochem. 2005, 95, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, W.; Reiser, G. Activation of protease-activated receptors in astrocytes evokes a novel neuroprotective pathway through release of chemokines of the growth-regulated oncogene/cytokine-induced neutrophil chemoattractant family. Eur. J. Neurosci. 2007, 26, 3159–3168. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Pyo, H.; Jou, I.; Joe, E. Thrombin induces NO release from cultured rat microglia via protein kinase C, mitogen-activated protein kinase, and NF-κB. J. Biol. Chem. 2000, 275, 29955–29959. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Q.; Zhou, Y.P.; Yang, H. Donepezil combined with natural hirudin improves the clinical symptoms of patients with mild-to-moderate Alzheimer’s disease: A 20-week open-label pilot study. Int. J. Med. Sci. 2012, 9, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Ikeda, K.; Kondo, H.; McGeer, P.L. Thrombin accumulation in brains of patients with Alzheimer’s disease. Neurosci. Lett. 1992, 146, 152–154. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Striggow, F.; Riek, M.; Breder, J.; Henrich-Noack, P.; Reymann, K.G.; Reiser, G. The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but causes degeneration at high concentrations. Proc. Natl. Acad. Sci. USA 2000, 97, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Mirante, O.; Price, M.; Puentes, W.; Castillo, X.; Benakis, C.; Thevenet, J.; Monard, D.; Hirt, L. Endogenous protease nexin-1 protects against cerebral ischemia. Int. J. Mol. Sci. 2013, 14, 16719–16731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olanow, C.W.; Tatton, W.G. Etiology and pathogenesis of Parkinson’s disease. Annu. Rev. Neurosci. 1999, 22, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Carreno-Muller, E.; Herrera, A.J.; de Pablos, R.M.; Tomás-Camardiel, M.; Venero, J.L.; Cano, J.; Machado, A. Thrombin induces in vivo degeneration of nigral dopaminergic neurones along with the activation of microglia. J. Neurochem. 2003, 84, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Nagai, A.; Kobayashi, S.; Kim, S.U. Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: Astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J. Neuropathol. Exp. Neurol. 2006, 65, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Katsuki, H.; Okawara, M.; Shibata, H.; Kume, T.; Akaike, A. Nitric oxide-producing microglia mediate thrombin-induced degeneration of dopaminergic neurons in rat midbrain slice culture. J. Neurochem. 2006, 97, 1232–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.Y.; Oh, Y.J.; Jin, B.K. Thrombin-activated microglia contribute to death of dopaminergic neurons in rat mesencephalic cultures: Dual roles of mitogen-activated protein kinase signaling pathways. Glia 2005, 51, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Lee, D.Y.; Ryu, J.K.; Kim, J.; Joe, E.H.; Jin, B.K. Thrombin induces nigral dopaminergic neurodegeneration in vivo by altering expression of death-related proteins. Neurobiol. Dis. 2003, 14, 181–193. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S36. [Google Scholar] [CrossRef] [PubMed]
- Lee da, Y.; Park, K.W.; Jin, B.K. Thrombin induces neurodegeneration and microglial activation in the cortex in vivo and in vitro: Proteolytic and non-proteolytic actions. Biochem. Biophys. Res. Commun. 2006, 346, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Keep, R.F.; Hua, Y.; Richardson, R.J.; Schallert, T.; Xi, G. Thrombin preconditioning provides protection in a 6-hydroxydopamine Parkinson’s disease model. Neurosci. Lett. 2005, 373, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Keep, R.F.; Schallert, T.; Hua, Y.; Richardson, R.J.; Xi, G. Protease-activated receptor-1 mediates protection elicited by thrombin preconditioning in a rat 6-hydroxydopamine model of Parkinson’s disease. Brain Res. 2006, 1116, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.J.; de Pablos, R.M.; Carreño-Müller, E.; Villarán, R.F.; Venero, J.L.; Tomás-Camardiel, M.; Cano, J.; Machado, A. The intrastriatal injection of thrombin in rat induced a retrograde apoptotic degeneration of nigral dopaminergic neurons through synaptic elimination. J. Neurochem. 2008, 105, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Hua, Y.; Richardson, R.J.; Xi, G.; Keep, R.F.; Schallert, T. The effect of thrombin on a 6-hydroxydopamine model of Parkinson’s disease depends on timing. Behav. Brain Res. 2007, 183, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Udaka, F.; Takahashi, M.; Nishinaka, K.; Kameyama, M. Secondary parkinsonism following midbrain hemorrhage. Clin. Neurol. 1997, 37, 266–269. [Google Scholar]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimer Dis. 2006, 9, 51–58. [Google Scholar]
- Pompili, E.; Nori, S.L.; Geloso, M.C.; Guadagni, E.; Corvino, V.; Michetti, F.; Fumagalli, L. Trimethyltin-induced differential expression of PAR subtypes in reactive astrocytes of the rat hippocampus. Brain Res. Mol. Brain Res. 2004, 122, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, P.J.; Su, J.; Cotman, C.W.; Cunningham, D.D. Protease nexin-1, a potent thrombin inhibitor, is reduced around cerebral blood vessels in Alzheimer’s disease. Brain Res. 1994, 668, 160–170. [Google Scholar] [CrossRef]
- Suo, Z.; Wu, M.; Citron, B.A.; Palazzo, R.E.; Festoff, B.W. Rapid tau aggregation and delayed hippocampal neuronal death induced by persistent thrombin signaling. J. Biol. Chem. 2003, 278, 37681–37689. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Thrombin causes cell spreading and redistribution of beta-amyloid immunoreactivity in cultured hippocampal neurons. J. Neurochem. 1996, 67, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Smith-Swintosky, V.L.; Zimmer, S.; Fenton, J.W., 2nd; Mattson, M.P. Opposing actions of thrombin and protease nexin-1 on amyloid β-peptide toxicity and on accumulation of peroxides and calcium in hippocampal neurons. J. Neurochem. 1995, 65, 1415–1418. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Vaughan, P.J.; Cunningham, D.D.; Cotman, C.W. Thrombin attenuates neuronal cell death and modulates astrocyte reactivity induced by β-amyloid in vitro. J. Neurochem. 1996, 66, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Lee, D.Y.; Kim, S.U.; Jin, B.K. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: Role of microglial NADPH oxidase. J. Neurosci. 2005, 25, 4082–4090. [Google Scholar] [CrossRef] [PubMed]
- Thirumangalakudi, L.; Samany, P.G.; Owoso, A.; Wiskar, B.; Grammas, P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimer Dis. 2006, 10, 111–118. [Google Scholar]
- Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J.; Martinez, J.; Grammas, P. Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 2013, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Van der Poll, T.; Büller, H.R.; ten Cate, H.; Wortel, C.H.; Bauer, K.A.; van Deventer, S.J.; Hack, C.E.; Sauerwein, H.P.; Rosenberg, R.D.; ten Cate, J.W. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N. Engl. J. Med. 1990, 322, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Paterson, P.Y.; Koh, C.S.; Kwaan, H.C. Role of the clotting system in the pathogenesis of neuroimmunologic disease. Fed. Proc. 1987, 46, 91–96. [Google Scholar] [PubMed]
- Kwon, E.E.; Prineas, J.W. Blood-brain barrier abnormalities in longstanding multiple sclerosis lesions. An immunohistochemical study. J. Neuropathol. Exp. Neurol. 1994, 53, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Ryu, J.K.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Bedard, C.; Hakozaki, H.; et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 2012, 3, 1227. [Google Scholar] [CrossRef] [PubMed]
- Boven, L.A.; Vergnolle, N.; Henry, S.D.; Silva, C.; Imai, Y.; Holden, J.; Warren, K.; Hollenberg, M.D.; Power, C. Up-regulation of proteinase-activated receptor 1 expression in astrocytes during HIV encephalitis. J. Immunol. 2003, 170, 2638–2646. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Ichikawa, M.; Inoue, A.; Itoh, M.; Kyogashima, M.; Sekiguchi, Y.; Nakamura, S.; Komiyama, A.; Koh, C. Plasma thrombin-antithrombin III complex is associated with the severity of experimental autoimmune encephalomyelitis. J. Neurol. Sci. 2001, 185, 89–93. [Google Scholar] [CrossRef]
- Davalos, D.; Baeten, K.M.; Whitney, M.A.; Mullins, E.S.; Friedman, B.; Olson, E.S.; Ryu, J.K.; Smirnoff, D.S.; Petersen, M.A.; Bedard, C.; et al. Early detection of thrombin activity in neuroinflammatory disease. Ann. Neurol. 2014, 75, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Richter-Landsberg, C.; Reiser, G. Expression of protease-activated receptors (PARs) in OLN-93 oligodendroglial cells and mechanism of PAR-1-induced calcium signaling. Neuroscience 2004, 126, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh, F.; Tsutsui, S.; Vergnolle, N.; Boven, L.A.; Shariat, N.; Vodjgani, M.; Warren, K.G.; Andrade-Gordon, P.; Hollenberg, M.D.; Power, C. Proteinase-activated receptor 2 modulates neuroinflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J. Exp. Med. 2006, 203, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Juhler, M.; Barry, D.I.; Offner, H.; Konat, G.; Klinken, L.; Paulson, O.B. Blood-brain and blood-spinal cord barrier permeability during the course of experimental allergic encephalomyelitis in the rat. Brain Res. 1984, 302, 347–355. [Google Scholar] [CrossRef]
- Kim, H.N.; Kim, Y.R.; Ahn, S.M.; Lee, S.K.; Shin, H.K.; Choi, B.T. Protease activated receptor-1 antagonist ameliorates the clinical symptoms of experimental autoimmune encephalomyelitis via inhibiting breakdown of blood brain barrier. J. Neurochem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh, F.; Vergnolle, N.; McArthur, J.C.; Silva, C.; Vodjgani, M.; Andrade-Gordon, P.; Hollenberg, M.D.; Power, C. Proteinase-activated receptor-2 induction by neuroinflammation prevents neuronal death during HIV infection. J. Immunol. 2005, 174, 7320–7329. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Zekas, E.; Lodge, R.; Susan-Resiga, D.; Marcinkiewicz, E.; Essalmani, R.; Mihara, K.; Ramachandran, R.; Asahchop, E.; Gelman, B.; et al. Neuroinflammation-induced interactions between protease-activated receptor 1 and proprotein convertases in HIV-associated neurocognitive disorder. Mol. Cell. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hurley, A.; Smith, M.; Karpova, T.; Hasley, R.B.; Belkina, N.; Shaw, S.; Balenga, N.; Druey, K.M.; Nickel, E.; Packard, B.; et al. Enhanced effector function of CD8(+) T cells from healthy controls and HIV-infected patients occurs through thrombin activation of protease-activated receptor 1. J. Infect. Dis. 2013, 207, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Xiao, P.; Usami, O.; Hattori, T. Thrombin activates envelope glycoproteins of HIV type 1 and enhances fusion. Microbes Infect. 2004, 6, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.V.; Brummel-Ziedins, K.; Neuhaus, J.; Duprez, D.; Cummins, N.; Dalmau, D.; DeHovitz, J.; Lehmann, C.; Sullivan, A.; Woolley, I.; et al. HIV replication alters the composition of extrinsic pathway coagulation factors and increases thrombin generation. J. Am. Heart Assoc. 2013, 2, e000264. [Google Scholar] [CrossRef] [PubMed]
- Ivey, N.S.; MacLean, A.G.; Lackner, A.A. Acquired immunodeficiency syndrome and the blood-brain barrier. J. Neurovirol. 2009, 15, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Hsue, P.Y.; Scherzer, R.; Grunfeld, C.; Nordstrom, S.M.; Schnell, A.; Kohl, L.P.; Nitta, E.; Martin, J.N.; Deeks, S.G.; Weiss, E.J. HIV infection is associated with decreased thrombin generation. Clin. Infect. Dis. 2012, 54, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.J.; Valdes-Sueiras, M.; Commins, D.L.; Yong, W.; Carlson, M. HIV stroke risk: Evidence and implications. Ther. Adv. Chronic Dis. 2013, 4, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.I.; Mendelow, A.D.; Hanley, D.F. Intracerebral haemorrhage. Lancet 2009, 373, 1632–1644. [Google Scholar] [CrossRef]
- Riek-Burchardt, M.; Striggow, F.; Henrich-Noack, P.; Reiser, G.; Reymann, K.G. Increase of prothrombin-mRNA after global cerebral ischemia in rats, with constant expression of protease nexin-1 and protease-activated receptors. Neurosci. Lett. 2002, 329, 181–184. [Google Scholar] [CrossRef]
- Thevenet, J.; Angelillo-Scherrer, A.; Price, M.; Hirt, L. Coagulation factor Xa activates thrombin in ischemic neural tissue. J. Neurochem. 2009, 111, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, T.; Henrich-Noack, P.; Sedehizade, F.; Goertler, M.; Wallesch, C.W.; Reymann, K.G.; Reiser, G. Transient focal ischemia in rat brain differentially regulates mRNA expression of protease-activated receptors 1 to 4. J. Neurosci. Res. 2004, 75, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Henrich-Noack, P.; Riek-Burchardt, M.; Baldauf, K.; Reiser, G.; Reymann, K.G. Focal ischemia induces expression of protease-activated receptor1 (PAR1) and PAR3 on microglia and enhances PAR4 labeling in the penumbra. Brain Res. 2006, 1070, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, W.; Li, J.Y.; Chai, B.X.; Peng, J.; Wang, H.; Mulholland, M.W. Induction of apoptosis by thrombin in the cultured neurons of dorsal motor nucleus of the vagus. Neurogastroenterol. Motil. 2011, 23, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.D.; Pulliam, L.; Vaughan, P.J. Protease nexin-1 and thrombin: Injury-related processes in the brain. Thromb. Haemost. 1993, 70, 168–171. [Google Scholar] [PubMed]
- Hoffmann, M.C.; Nitsch, C.; Scotti, A.L.; Reinhard, E.; Monard, D. The prolonged presence of glia-derived nexin, an endogenous protease inhibitor, in the hippocampus after ischemia-induced delayed neuronal death. Neuroscience 1992, 49, 397–408. [Google Scholar] [CrossRef]
- Xi, G.; Keep, R.F.; Hua, Y.; Xiang, J.; Hoff, J.T. Attenuation of thrombin-induced brain edema by cerebral thrombin preconditioning. Stroke 1999, 30, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Henrich-Noack, P.; Striggow, F.; Reiser, G.; Reymann, K.G. Preconditioning with thrombin can be protective or worsen damage after endothelin-1-induced focal ischemia in rats. J. Neurosci. Res. 2006, 83, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Masada, T.; Xi, G.; Hua, Y.; Keep, R.F. The effects of thrombin preconditioning on focal cerebral ischemia in rats. Brain Res. 2000, 867, 173–179. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, J.; Hua, Y.; Keep, R.F.; Xiang, J.; Hoff, J.T.; Xi, G. Thrombin-receptor activation and thrombin-induced brain tolerance. J. Cereb. Blood Flow Metab. 2002, 22, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Karabiyikoglu, M.; Hua, Y.; Keep, R.F.; Ennis, S.R.; Xi, G. Intracerebral hirudin injection attenuates ischemic damage and neurologic deficits without altering local cerebral blood flow. J. Cereb. Blood Flow Metab. 2004, 24, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Granziera, C.; Thevenet, J.; Price, M.; Wiegler, K.; Magistretti, P.J.; Badaut, J.; Hirt, L. Thrombin-induced ischemic tolerance is prevented by inhibiting c-jun N-terminal kinase. Brain Res. 2007, 1148, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Hua, Y.; Nakamura, T.; Keep, R.F.; Xi, G. Up-regulation of brain ceruloplasmin in thrombin preconditioning. Acta Neurochir. Suppl. 2006, 96, 203–206. [Google Scholar] [PubMed]
- Wang, Y.; Luo, W.; Stricker, R.; Reiser, G. Protease-activated receptor-1 protects rat astrocytes from apoptotic cell death via JNK-mediated release of the chemokine GRO/CINC-1. J. Neurochem. 2006, 98, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Reiser, G.; Keep, R.F. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: Deleterious or protective? J. Neurochem. 2003, 84, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.; Katsuki, H.; Kume, T.; Akaike, A. Thrombin-induced delayed injury involves multiple and distinct signaling pathways in the cerebral cortex and the striatum in organotypic slice cultures. Neurobiol. Dis. 2006, 22, 130–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Keep, R.F.; Schallert, T.; Hoff, J.T.; Xi, G. A thrombin inhibitor reduces brain edema, glioma mass and neurological deficits in a rat glioma model. Acta Neurochir. Suppl. 2003, 86, 503–506. [Google Scholar] [PubMed]
- Hua, Y.; Wu, J.; Keep, R.F.; Hoff, J.T.; Xi, G. Thrombin exacerbates brain edema in focal cerebral ischemia. Acta Neurochir. Suppl. 2003, 86, 163–166. [Google Scholar] [PubMed]
- Hua, Y.; Wu, J.; Keep, R.F.; Nakamura, T.; Hoff, J.T.; Xi, G. Tumor necrosis factor-alpha increases in the brain after intracerebral hemorrhage and thrombin stimulation. Neurosurgery 2006, 58, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, G.; Dirnagl, U. Neuroprotective role of astrocytes in cerebral ischemia: Focus on ischemic preconditioning. Glia 2005, 50, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R.; Camerer, E. PARticipation in inflammation. J. Clin. Investig. 2003, 111, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Masada, T.; Hua, Y.; Xi, G.; Yang, G.Y.; Hoff, J.T.; Keep, R.F.; Nagao, S. Overexpression of interleukin-1 receptor antagonist reduces brain edema induced by intracerebral hemorrhage and thrombin. Acta Neurochir. Suppl. 2003, 86, 463–467. [Google Scholar] [PubMed]
- Nakamura, T.; Xi, G.; Park, J.W.; Hua, Y.; Hoff, J.T.; Keep, R.F. Holo-transferrin and thrombin can interact to cause brain damage. Stroke 2005, 36, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Keep, R.F.; Hua, Y.; Hoff, J.T. Thrombin preconditioning, heat shock proteins and thrombin-induced brain edema. Acta Neurochir. Suppl. 2000, 76, 511–515. [Google Scholar] [PubMed]
- Hua, Y.; Keep, R.F.; Hoff, J.T.; Xi, G. Brain injury after intracerebral hemorrhage: The role of thrombin and iron. Stroke 2007, 38, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Svedin, P.; Hagberg, H.; Savman, K.; Zhu, C.; Mallard, C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J. Neurosci. 2007, 27, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Hollenberg, M.D.; Yong, V.W. Combination of thrombin and matrix metalloproteinase-9 exacerbates neurotoxicity in cell culture and intracerebral hemorrhage in mice. J. Neurosci. 2006, 26, 10281–10291. [Google Scholar] [CrossRef] [PubMed]
- Junge, C.E.; Sugawara, T.; Mannaioni, G.; Alagarsamy, S.; Conn, P.J.; Brat, D.J.; Chan, P.H.; Traynelis, S.F. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2003, 100, 13019–13024. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.E.; Lyuboslavsky, P.; Traynelis, S.F.; McKeon, R.J. PAR-1 deficiency protects against neuronal damage and neurologic deficits after unilateral cerebral hypoxia/ischemia. J. Cereb. Blood Flow Metab. 2004, 24, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Mandujano, G.; Vazquez-Juarez, E.; Hernandez-Benitez, R.; Pasantes-Morales, H. Thrombin potently enhances swelling-sensitive glutamate efflux from cultured astrocytes. Glia 2007, 55, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Zukin, R.S. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007, 30, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.; Weiss, J.H. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr. Opin. Neurobiol. 2006, 16, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Gorbacheva, L.R.; Storozhevykh, T.P.; Pinelis, V.G.; Ishiwata, S.; Strukova, S.M. Modulation of hippocampal neuron survival by thrombin and factor Xa. Biochem. Biokhimiia 2006, 71, 1082–1089. [Google Scholar] [CrossRef]
- Putcha, G.V.; Moulder, K.L.; Golden, J.P.; Bouillet, P.; Adams, J.A.; Strasser, A.; Johnson, E.M. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 2001, 29, 615–628. [Google Scholar] [CrossRef]
- Rao, H.V.; Thirumangalakudi, L.; Desmond, P.; Grammas, P. Cyclin D1, cdk4, and Bim are involved in thrombin-induced apoptosis in cultured cortical neurons. J. Neurochem. 2007, 101, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, J.; Iyirhiaro, G.; Aleyasin, H.; Rios, M.; Vincent, I.; Callaghan, S.; Bland, R.J.; Slack, R.S.; During, M.J.; Park, D.S. Multiple cyclin-dependent kinases signals are critical mediators of ischemia/hypoxic neuronal death in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 14080–14085. [Google Scholar] [CrossRef] [PubMed]
- Nicole, O.; Goldshmidt, A.; Hamill, C.E.; Sorensen, S.D.; Sastre, A.; Lyuboslavsky, P.; Hepler, J.R.; McKeon, R.J.; Traynelis, S.F. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J. Neurosci. 2005, 25, 4319–4329. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Katsuki, H.; Fujimoto, S.; Takagi, M.; Kume, T.; Akaike, A. Involvement of thrombin and mitogen-activated protein kinase pathways in hemorrhagic brain injury. Exp. Neurol. 2007, 206, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markwardt, F. Hirudin as alternative anticoagulant—A historical review. Semin. Thromb. Hemost. 2002, 28, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Ten Cate, H. Challenging the anticoagulant paradigm? J. Thromb. Haemost. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sugg, R.M.; Pary, J.K.; Uchino, K.; Baraniuk, S.; Shaltoni, H.M.; Gonzales, N.R.; Mikulik, R.; Garami, Z.; Shaw, S.G.; Matherne, D.E.; et al. Argatroban tPA stroke study: Study design and results in the first treated cohort. Arch. Neurol. 2006, 63, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Barreto, A.D.; Alexandrov, A.V.; Lyden, P.; Lee, J.; Martin-Schild, S.; Shen, L.; Wu, T.C.; Sisson, A.; Pandurengan, R.; Chen, Z. The argatroban and tissue-type plasminogen activator stroke study: Final results of a pilot safety study. Stroke 2012, 43, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Lyden, P.; Pereira, B.; Chen, B.; Zhao, L.; Lamb, J.; Lei, I.F.; Rajput, P. Direct thrombin inhibitor argatroban reduces stroke damage in 2 different models. Stroke 2014, 45, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, H.; Hamada, R. Role of thrombin in CNS damage associated with intracerebral haemorrhage: Opportunity for pharmacological intervention? CNS Drugs 2002, 16, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Kameda, K.; Kikkawa, Y.; Hirano, M.; Matsuo, S.; Sasaki, T.; Hirano, K. Combined argatroban and anti-oxidative agents prevents increased vascular contractility to thrombin and other ligands after subarachnoid haemorrhage. Br. J. Pharmacol. 2012, 165, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Jadhav, V.; Ayer, R.; Chen, W.; Suzuki, H.; Zhang, J.H. Thrombin inhibition by argatroban ameliorates early brain injury and improves neurological outcomes after experimental subarachnoid hemorrhage in rats. Stroke 2009, 40, 1530–1532. [Google Scholar] [CrossRef] [PubMed]
- Mhatre, M.; Hensley, K.; Nguyen, A.; Grammas, P. Chronic thrombin exposure results in an increase in apolipoprotein-E levels. J. Neurosci. Res. 2006, 84, 444–449. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Database. CID=216210. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/216210 (accessed on 23 November 2015).
- Eisert, W.G.; Hauel, N.; Stangier, J.; Wienen, W.; Clemens, A.; van Ryn, J. Dabigatran: An oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1885–1889. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krenzlin, H.; Lorenz, V.; Danckwardt, S.; Kempski, O.; Alessandri, B. The Importance of Thrombin in Cerebral Injury and Disease. Int. J. Mol. Sci. 2016, 17, 84. https://doi.org/10.3390/ijms17010084
Krenzlin H, Lorenz V, Danckwardt S, Kempski O, Alessandri B. The Importance of Thrombin in Cerebral Injury and Disease. International Journal of Molecular Sciences. 2016; 17(1):84. https://doi.org/10.3390/ijms17010084
Chicago/Turabian StyleKrenzlin, Harald, Viola Lorenz, Sven Danckwardt, Oliver Kempski, and Beat Alessandri. 2016. "The Importance of Thrombin in Cerebral Injury and Disease" International Journal of Molecular Sciences 17, no. 1: 84. https://doi.org/10.3390/ijms17010084