
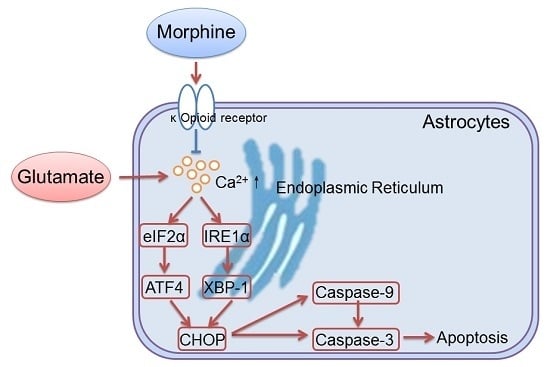
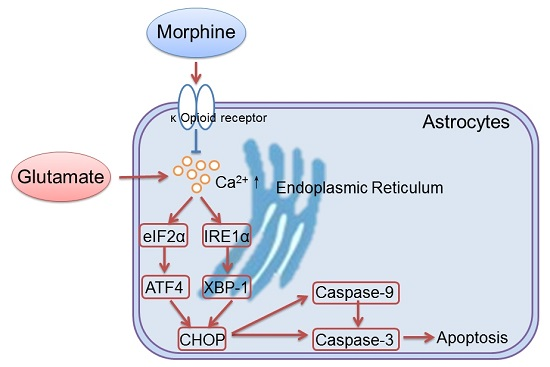
Morphine Protects Spinal Cord Astrocytes from Glutamate-Induced Apoptosis via Reducing Endoplasmic Reticulum Stress

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
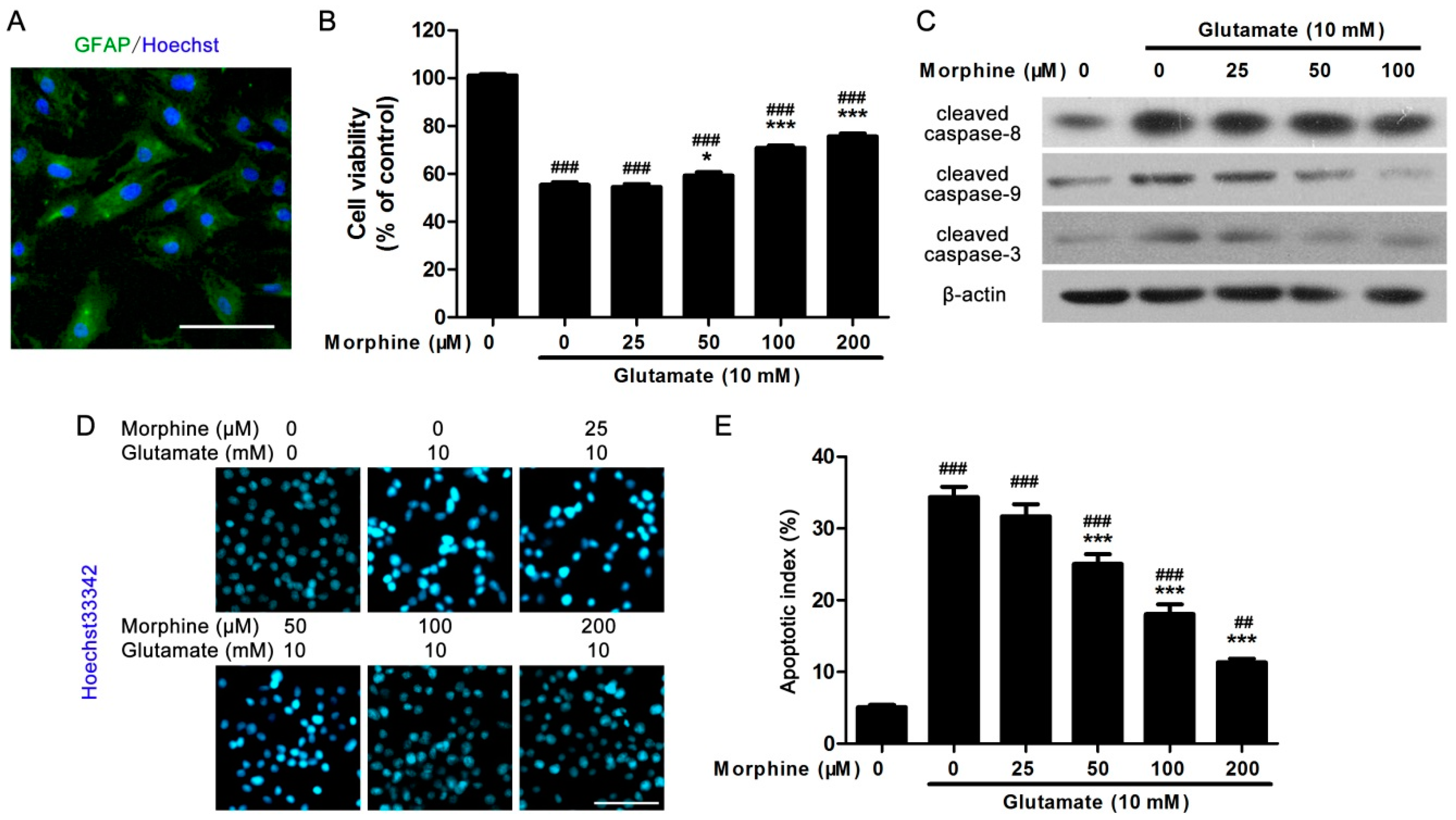
2.1. Morphine Protected Primary Cultured Spinal Cord Astrocytes from Glutamate-Induced Mitochondrial Apoptosis
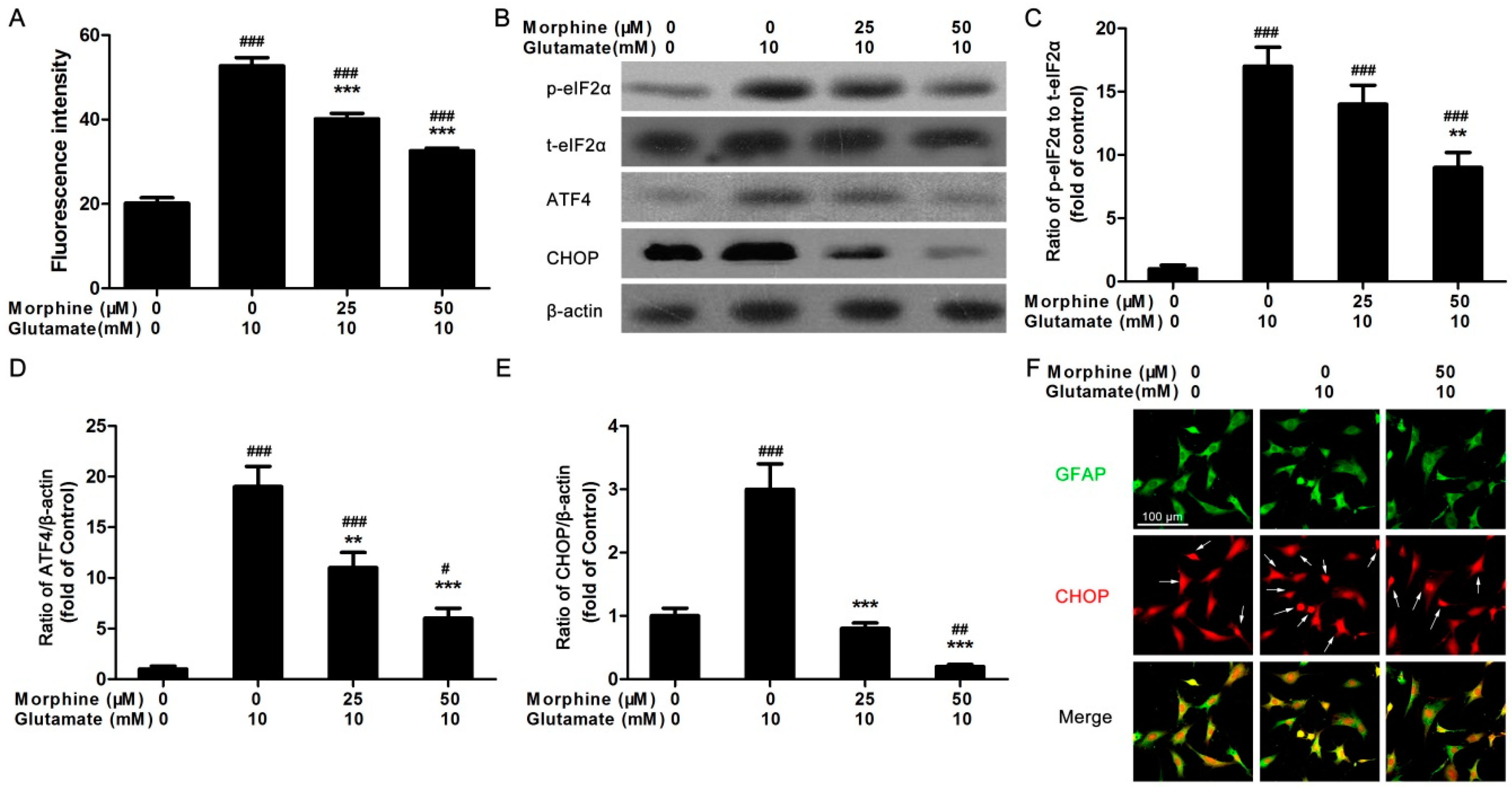
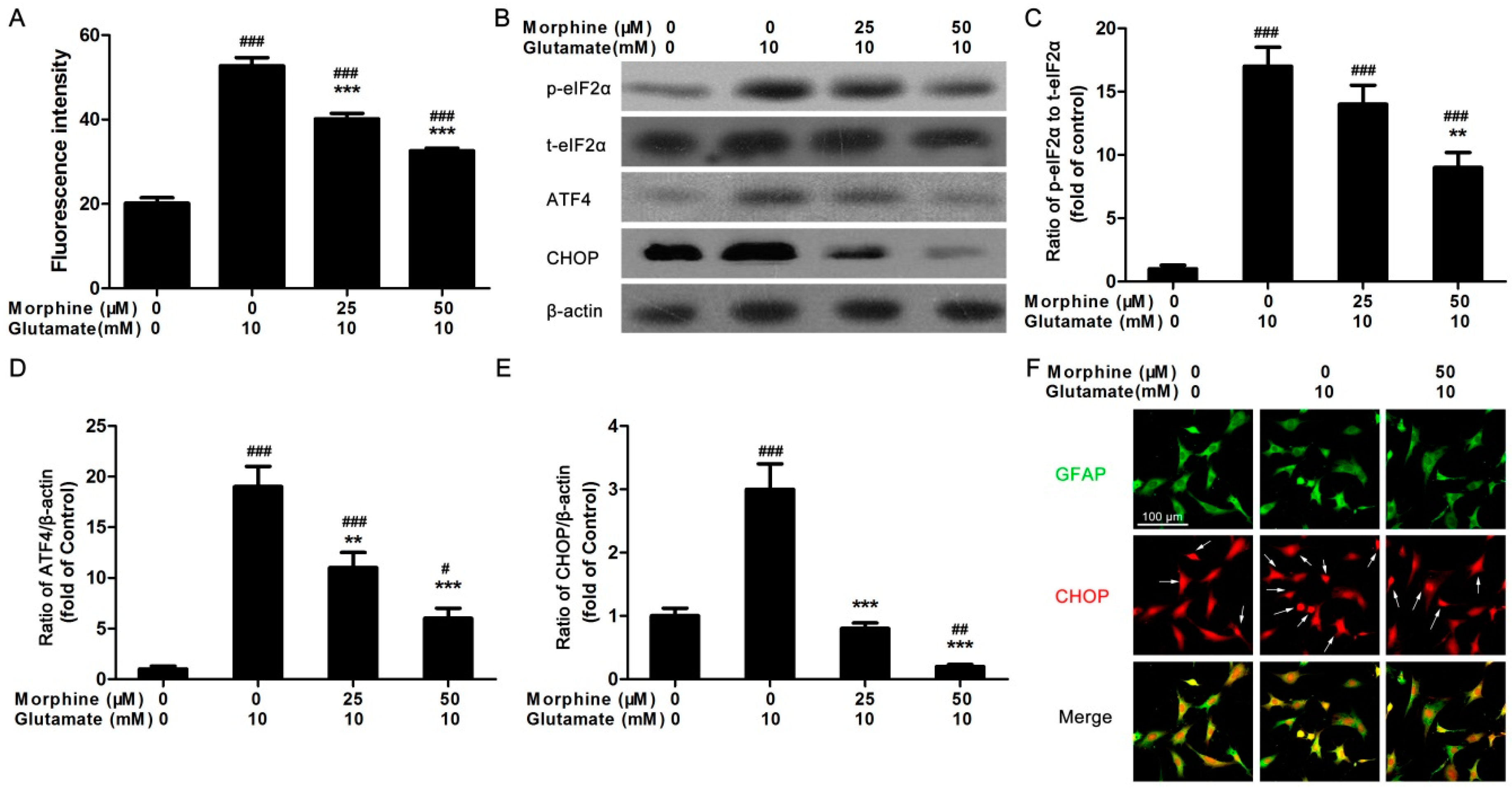
2.2. Morphine Reduced Glutamate-Induced Ca2+ Release and Endoplasmic Reticulum (ER) Stress
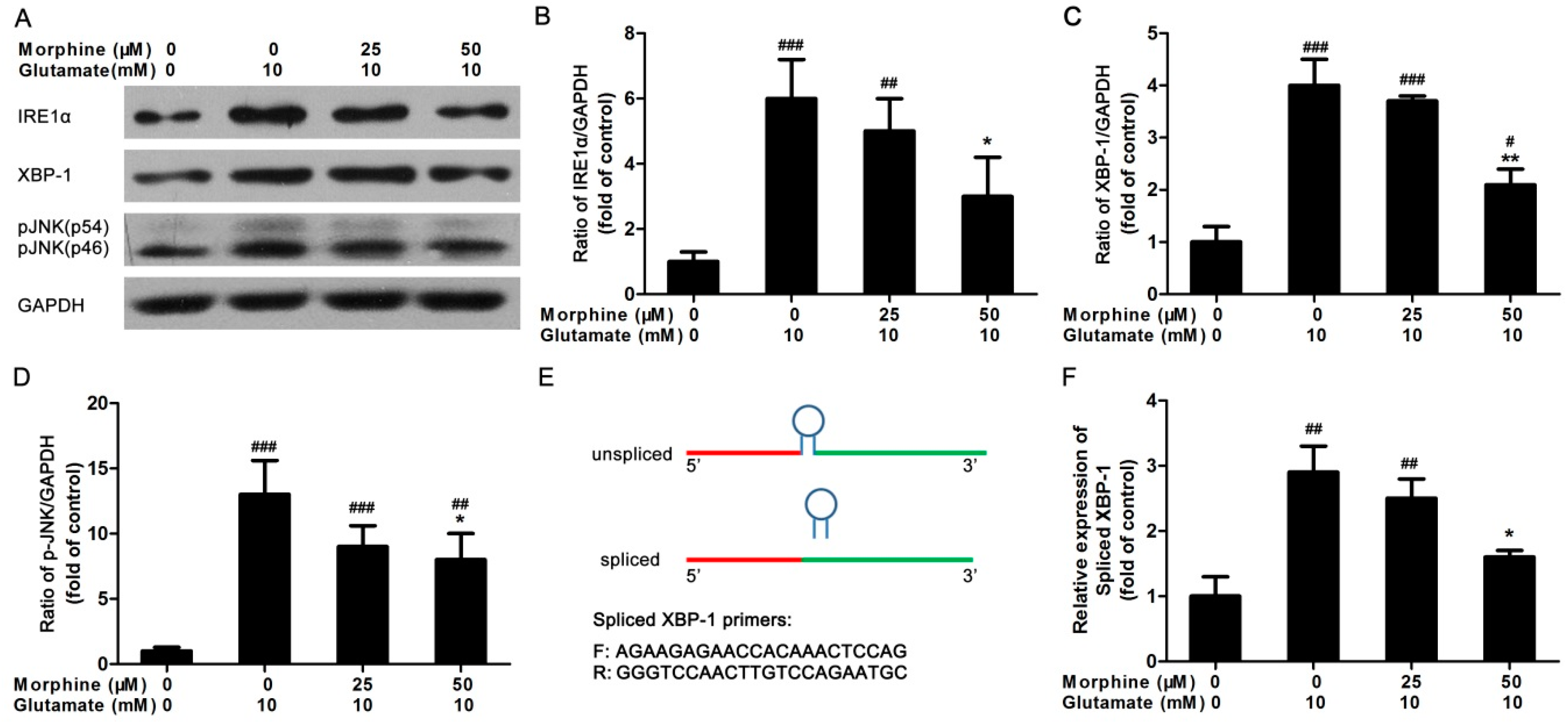
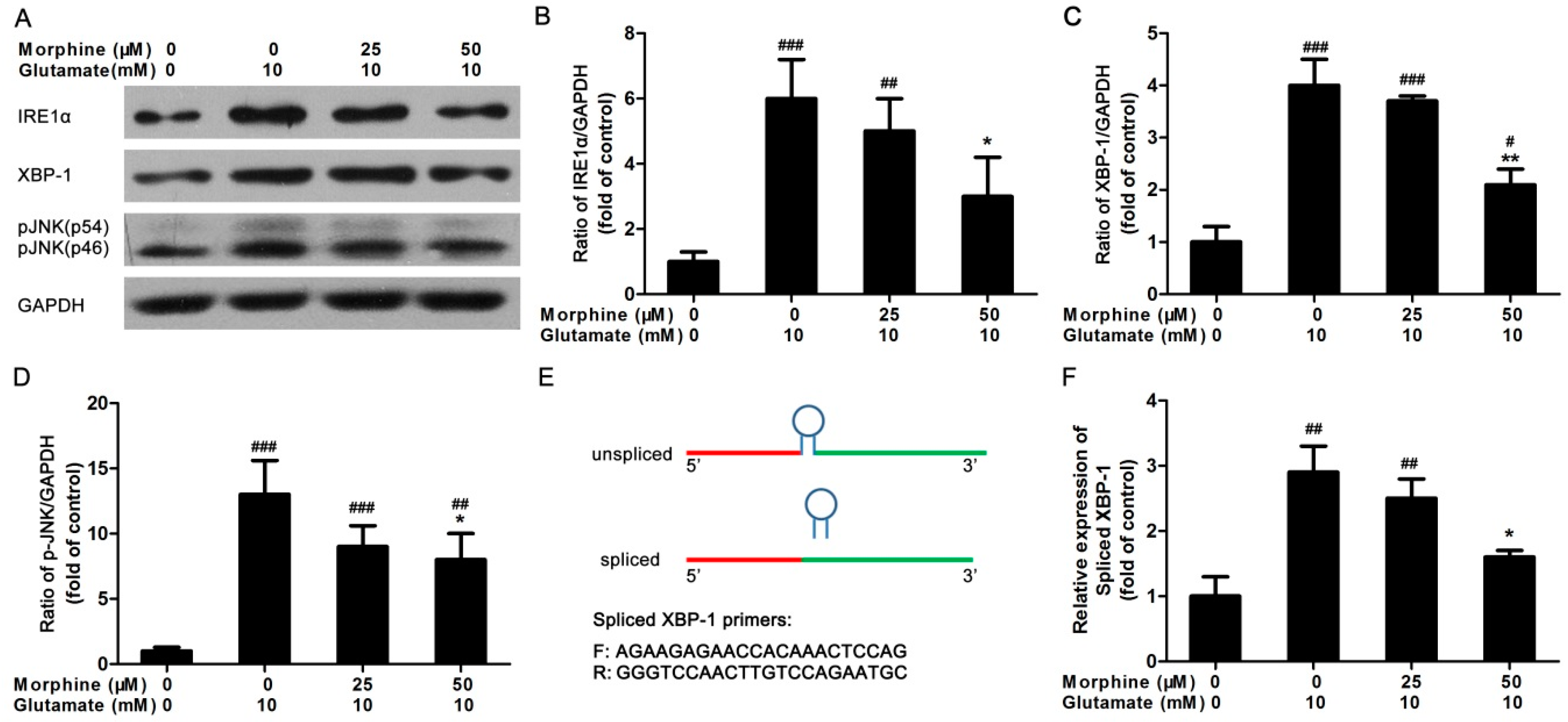
2.3. Inositol Requiring Kinase 1 (IRE1) Pathway Is Partially Involved in the Effect of Morphine on Glutamate-Treated Astrocytes
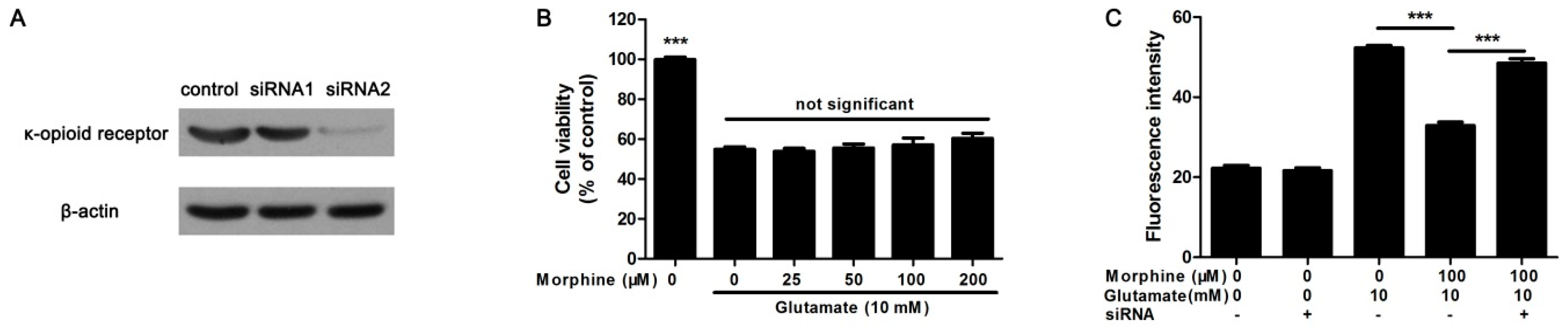
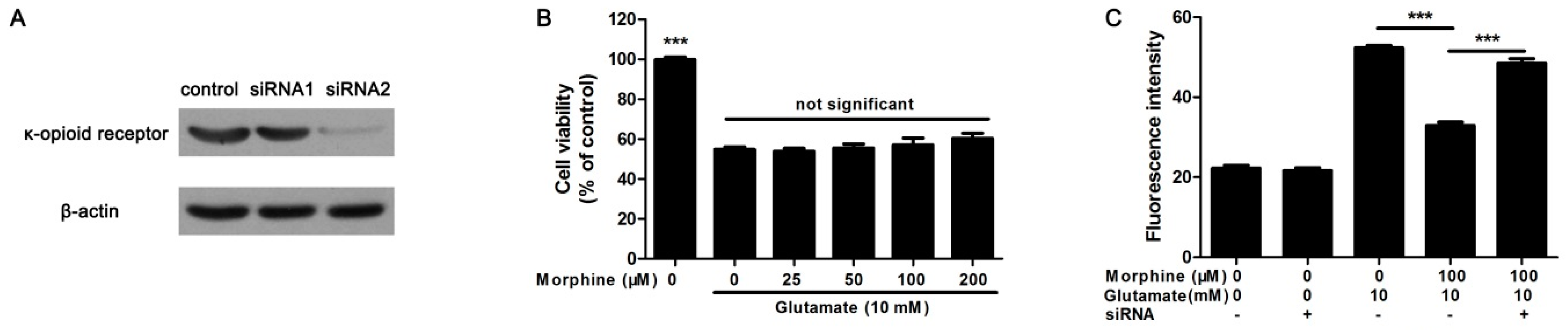
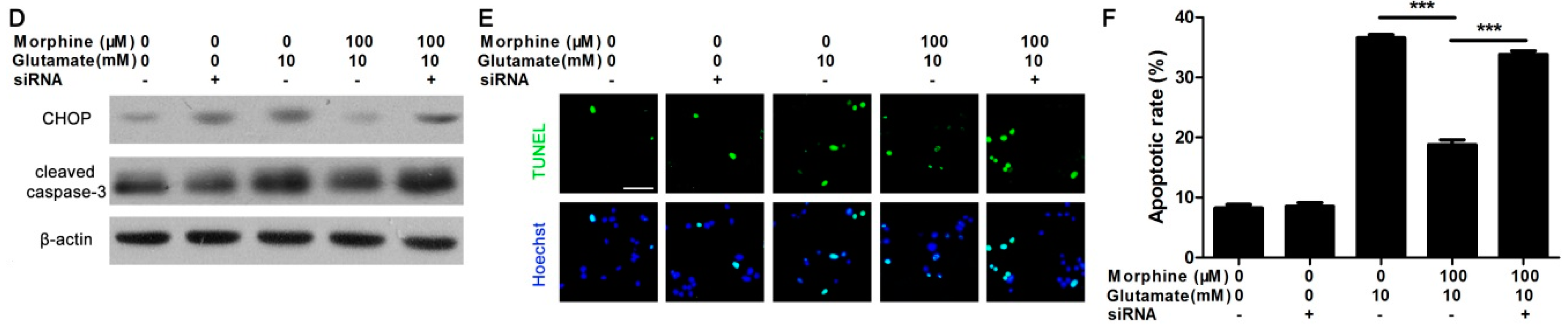
2.4. Knockdown κ-Receptor Suppressed the Protective Effect of Morphine on Glutamate-Stimulated Astrocytes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability Assay
4.3. Western Blot Analysis
4.4. Real-Time PCR Assay
4.5. Hoechst 33342 Staining
4.6. Calcium Mobilization Assay
4.7. Immunofluorescence
4.8. TUNEL Staining
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Glowinski, J.; Marin, P.; Tence, M.; Stella, N.; Giaume, C.; Premont, J. Glial receptors and their intervention in astrocyto-astrocytic and astrocyto-neuronal interactions. Glia 1994, 11, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castano, P.; Salido, G.M.; Gonzalez, A. Ethanol reduces kainate-evoked glutamate secretion in rat hippocampal astrocytes. Brain Res. 2011, 1402, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D. Reactive glia as rivals in regulating neuronal survival. Glia 1993, 7, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C.; Furness, D.N.; Zhou, Y. Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem. Int. 2016, 98, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Koriauli, S.; Natsvlishvili, N.; Barbakadze, T.; Mikeladze, D. Knockdown of interleukin-10 induces the redistribution of sigma1-receptor and increases the glutamate-dependent NADPH-oxidase activity in mouse brain neurons. Biol. Res. 2015, 48, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Al-Aref, R.; Zhao, D.; Shen, J.; Yan, Y.; Gao, Y. Astrocyte-conditioned medium attenuates glutamate-induced apoptotic cell death in primary cultured spinal cord neurons of rats. Neurol. Res. 2015, 37, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Tseng, E.E.; Brock, M.V.; Lange, M.S.; Troncoso, J.C.; Blue, M.E.; Lowenstein, C.J.; Johnston, M.V.; Baumgartner, W.A. Glutamate excitotoxicity mediates neuronal apoptosis after hypothermic circulatory arrest. Ann. Thorac. Surg. 2010, 89, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Parfenova, H.; Basuroy, S.; Bhattacharya, S.; Tcheranova, D.; Qu, Y.; Regan, R.F.; Leffler, C.W. Glutamate induces oxidative stress and apoptosis in cerebral vascular endothelial cells: Contributions of HO-1 and HO-2 to cytoprotection. Am. J. Physiol. Cell Physiol. 2006, 290, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.T.; Lefkowitz, R.J.; Caron, M.G. Mu-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [PubMed]
- Chakrabarti, S.; Wang, L.; Tang, W.J.; Gintzler, A.R. Chronic morphine augments adenylyl cyclase phosphorylation: Relevance to altered signaling during tolerance/dependence. Mol. Pharmacol. 1998, 54, 949–953. [Google Scholar] [PubMed]
- Martini, L.; Whistler, J.L. The role of μ opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr. Opin. Neurobiol. 2007, 17, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Deb, I.; Das, S. Thyroid hormones protect astrocytes from morphine-induced apoptosis by regulating nitric oxide and pERK 1/2 pathways. Neurochem. Int. 2011, 58, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Cheong, Y.P.; So, H.S.; Lee, K.M.; Kim, T.Y.; Oh, J.; Chung, Y.T.; Son, Y.; Kim, B.R.; Park, R. Protective effects of morphine in peroxynitrite-induced apoptosis of primary rat neonatal astrocytes: Potential involvement of G protein and phosphatidylinositol 3-kinase (PI3 kinase). Biochem. Pharmacol. 2001, 61, 779–786. [Google Scholar] [CrossRef]
- Lee, J.; Kim, M.S.; Park, C.; Jung, E.B.; Choi, D.H.; Kim, T.Y.; Moon, S.K.; Park, R. Morphine prevents glutamate-induced death of primary rat neonatal astrocytes through modulation of intracellular redox. Immunopharmacol. Immunotoxicol. 2004, 26, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Chao, Z.; Shanxi Medical University, Taiyuan. Unpublished work. 2016.
- Suwanjang, W.; Holmstrom, K.M.; Chetsawang, B.; Abramov, A.Y. Glucocorticoids reduce intracellular calcium concentration and protects neurons against glutamate toxicity. Cell Calcium 2013, 53, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Suarez, E.; Caldwell, A.L.; Allen, N.J. Role of astrocyte-synapse interactions in CNS disorders. J. Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Acioglu, C.; Mirabelli, E.; Baykal, A.T.; Ni, L.; Ratnayake, A.; Heary, R.F.; Elkabes, S. Toll like receptor 9 antagonism modulates spinal cord neuronal function and survival: Direct versus astrocyte-mediated mechanisms. Brain Behav. Immun. 2016, 56, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Meisingset, T.W.; Risa, O.; Brenner, M.; Messing, A.; Sonnewald, U. Alteration of glial-neuronal metabolic interactions in a mouse model of alexander disease. Glia 2010, 58, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Goursaud, S.; Maloteaux, J.M.; Hermans, E. Distinct expression and regulation of the glutamate transporter isoforms GLT-1a and GLT-1b in cultured astrocytes from a rat model of amyotrophic lateral sclerosis (hSOD1G93A). Neurochem. Int. 2009, 55, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wu, H.; Zeng, C.; Xiong, X.; Tang, S.; Tang, Z.; Sun, X. Apolipoprotein E protects astrocytes from hypoxia and glutamate-induced apoptosis. FEBS Lett. 2013, 587, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Szydlowska, K.; Gozdz, A.; Dabrowski, M.; Zawadzka, M.; Kaminska, B. Prolonged activation of ERK triggers glutamate-induced apoptosis of astrocytes: Neuroprotective effect of FK506. J. Neurochem. 2010, 113, 904–918. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Waagepetersen, H.S. Role of astrocytes in glutamate homeostasis: Implications for excitotoxicity. Neurotox. Res. 2005, 8, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Benjelloun, N.; Joly, L.M.; Palmier, B.; Plotkine, M.; Charriaut-Marlangue, C. Apoptotic mitochondrial pathway in neurones and astrocytes after neonatal hypoxia-ischaemia in the rat brain. Neuropathol. Appl. Neurobiol. 2003, 29, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Jiang, X.; Du, F.; Wang, X. PHAPI, CAS, and Hsp70 promote apoptosome formation by preventing Apaf-1 aggregation and enhancing nucleotide exchange on Apaf-1. Mol. Cell 2008, 30, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Nunez, V.; Jimenez Gonzalez, A.; Barreto-Valer, K.; Rodriguez, R.E. In vivo regulation of the μ opioid receptor: Role of the endogenous opioid agents. Mol. Med. 2013, 19, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional calcium and glutamate signaling in striatal astrocytes from huntington’s disease model mice. J. Neurosci. 2016, 36, 3453–3470. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.K.; McCarthy, K.D. Mature hippocampal astrocytes exhibit functional metabotropic and ionotropic glutamate receptors in situ. Glia 1999, 26, 1–11. [Google Scholar] [CrossRef]
- Warren, D.E.; Bickler, P.E.; Clark, J.P.; Gregersen, M.; Brosnan, H.; McKleroy, W.; Gabatto, P. Hypothermia and rewarming injury in hippocampal neurons involve intracellular Ca2+ and glutamate excitotoxicity. Neuroscience 2012, 207, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.S.; Hansson, E.; Ronnback, L. δ and κ opiate receptors in primary astroglial cultures. Part II: Receptor sets in cultures from various brain regions and interactions with β-receptor activated cyclic AMP. Neurochem. Res. 1992, 17, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Huang, C.C.; Lu, D.Y.; Wong, K.L.; Chen, Y.R.; Cheng, T.H.; Leung, Y.M. Ca2+ store depletion and endoplasmic reticulum stress are involved in P2X7 receptor-mediated neurotoxicity in differentiated NG108–15 cells. J. Cell. Biochem. 2012, 113, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tan, H.; Gu, Z.; Liu, Z.; Geng, Y.; Liu, Y.; Tong, H.; Tang, Y.; Qiu, J.; Su, L. Heat stress induces apoptosis through a Ca2+-mediated mitochondrial apoptotic pathway in human umbilical vein endothelial cells. PLoS ONE 2014, 9, e111083. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Ladriere, L.; Ortis, F.; Igoillo-Esteve, M.; Gurzov, E.N.; Lupi, R.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Glucagon-like peptide-1 agonists protect pancreatic β-cells from lipotoxic endoplasmic reticulum stress through upregulation of BiP and JunB. Diabetes 2009, 58, 2851–2862. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of chop-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Zhao, Z.; Cai, T.; Yang, P.; Cheng, M. Liraglutide alleviates diabetic cardiomyopathy by blocking CHOP-triggered apoptosis via the inhibition of the IRE-α pathway. Mol. Med. Rep. 2014, 9, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sheng, W.S.; Lokensgard, J.R.; Peterson, P.K. Morphine induces apoptosis of human microglia and neurons. Neuropharmacology 2002, 42, 829–836. [Google Scholar] [CrossRef]
- Yamada, H.; Shimoyama, N.; Sora, I.; Uhl, G.R.; Fukuda, Y.; Moriya, H.; Shimoyama, M. Morphine can produce analgesia via spinal κ opioid receptors in the absence of μ opioid receptors. Brain Res. 2006, 1083, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Du, L. PERK pathway is involved in oxygen-glucose-serum deprivation-induced NF-κB activation via ROS generation in spinal cord astrocytes. Biochem. Biophys. Res. Commun. 2015, 467, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhai, L.; Xue, R.; Shi, J.; Zeng, Q.; Gao, C. Mast cell tryptase contributes to pancreatic cancer growth through promoting angiogenesis via activation of angiopoietin-1. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Wang, C.; Ren, J.; Guo, X.; Yun, K. Morphine Protects Spinal Cord Astrocytes from Glutamate-Induced Apoptosis via Reducing Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2016, 17, 1523. https://doi.org/10.3390/ijms17101523
Zhang C, Wang C, Ren J, Guo X, Yun K. Morphine Protects Spinal Cord Astrocytes from Glutamate-Induced Apoptosis via Reducing Endoplasmic Reticulum Stress. International Journal of Molecular Sciences. 2016; 17(10):1523. https://doi.org/10.3390/ijms17101523
Chicago/Turabian StyleZhang, Chao, Chendan Wang, Jianbo Ren, Xiangjie Guo, and Keming Yun. 2016. "Morphine Protects Spinal Cord Astrocytes from Glutamate-Induced Apoptosis via Reducing Endoplasmic Reticulum Stress" International Journal of Molecular Sciences 17, no. 10: 1523. https://doi.org/10.3390/ijms17101523
APA StyleZhang, C., Wang, C., Ren, J., Guo, X., & Yun, K. (2016). Morphine Protects Spinal Cord Astrocytes from Glutamate-Induced Apoptosis via Reducing Endoplasmic Reticulum Stress. International Journal of Molecular Sciences, 17(10), 1523. https://doi.org/10.3390/ijms17101523