
Essential Roles of Natural Products and Gaseous Mediators on Neuronal Cell Death or Survival


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neuroprotection by Coffee Polyphenols
2.1. Coffee Consumption and Health
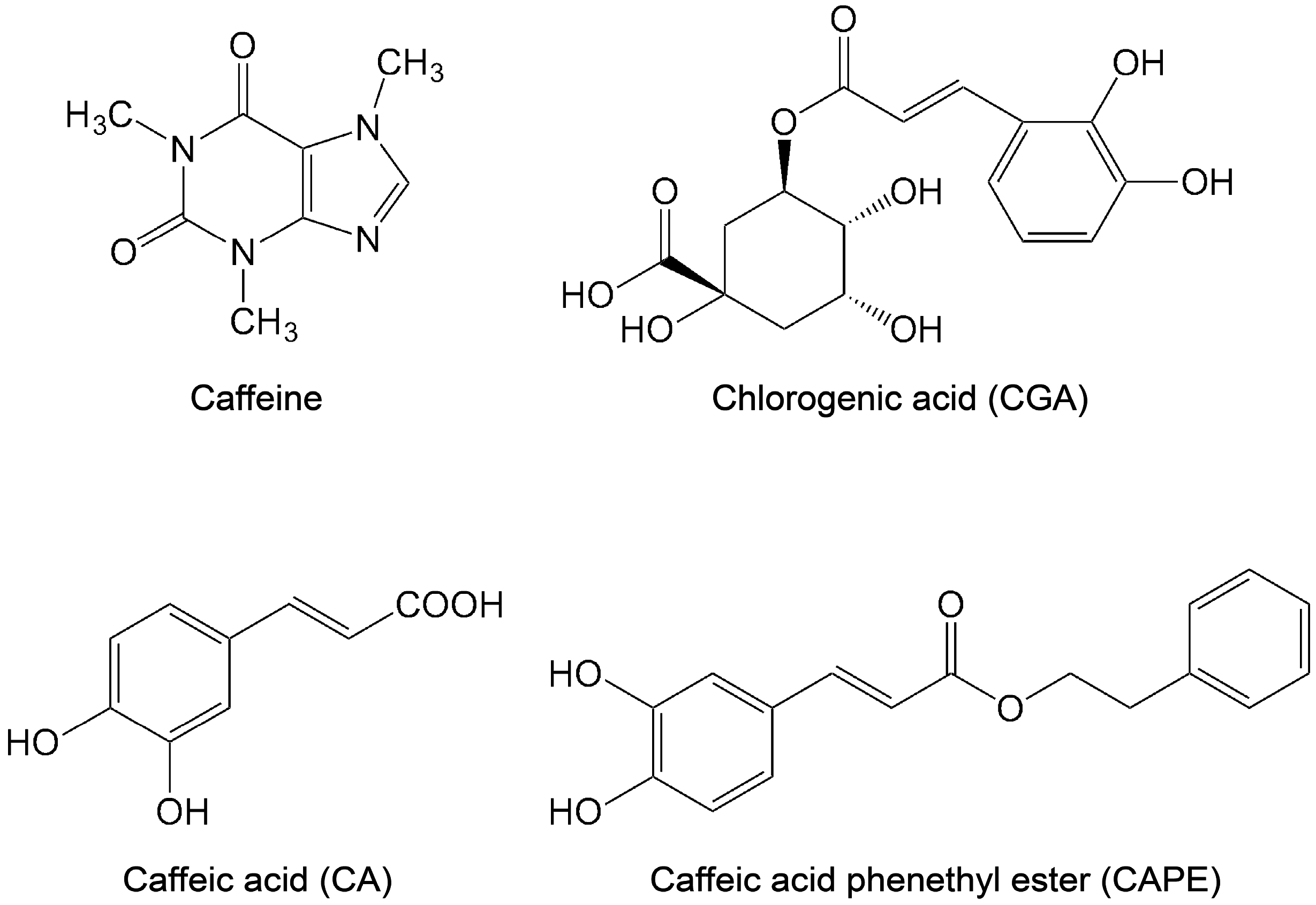
2.2. Caffeic Acid
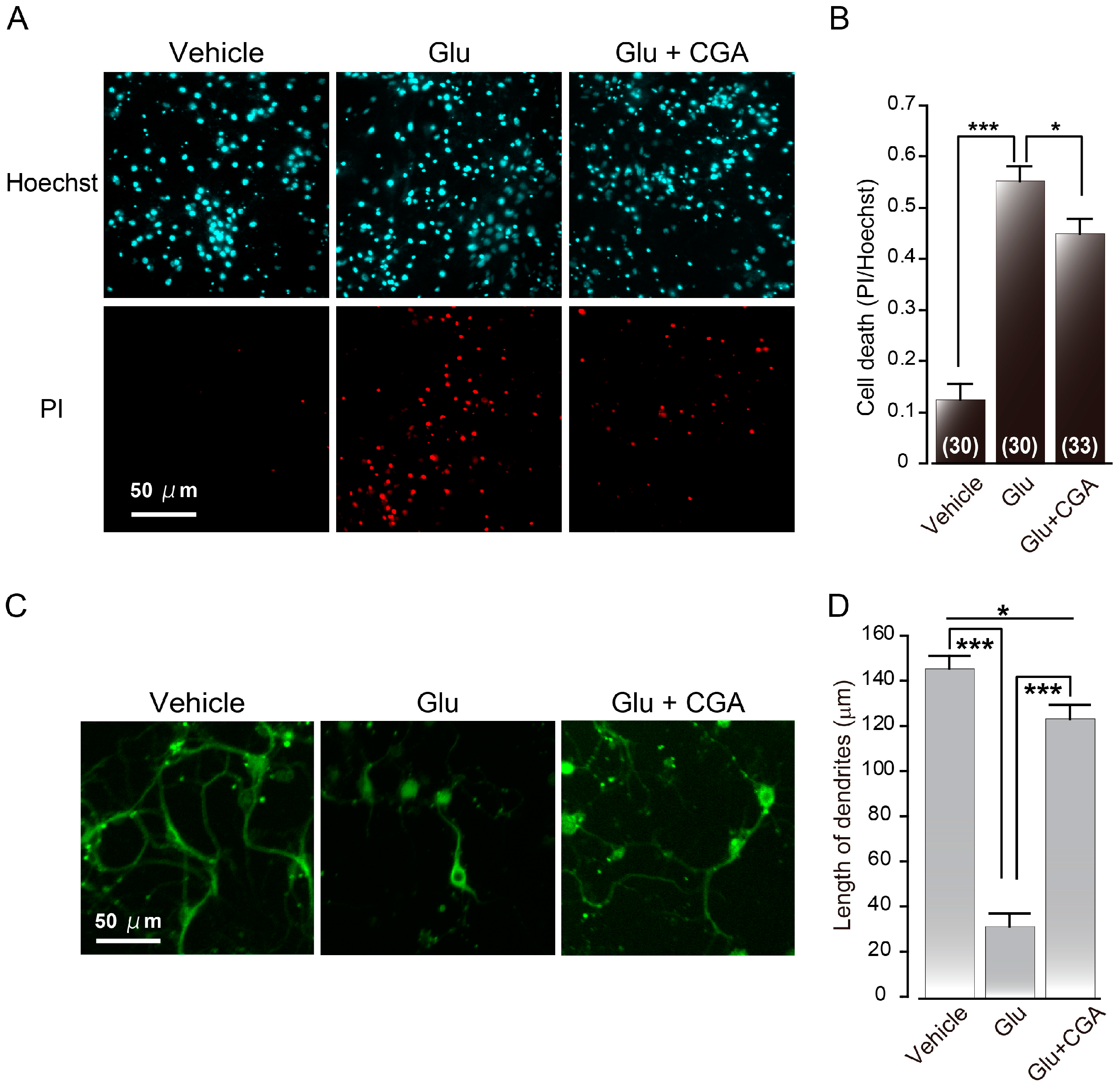
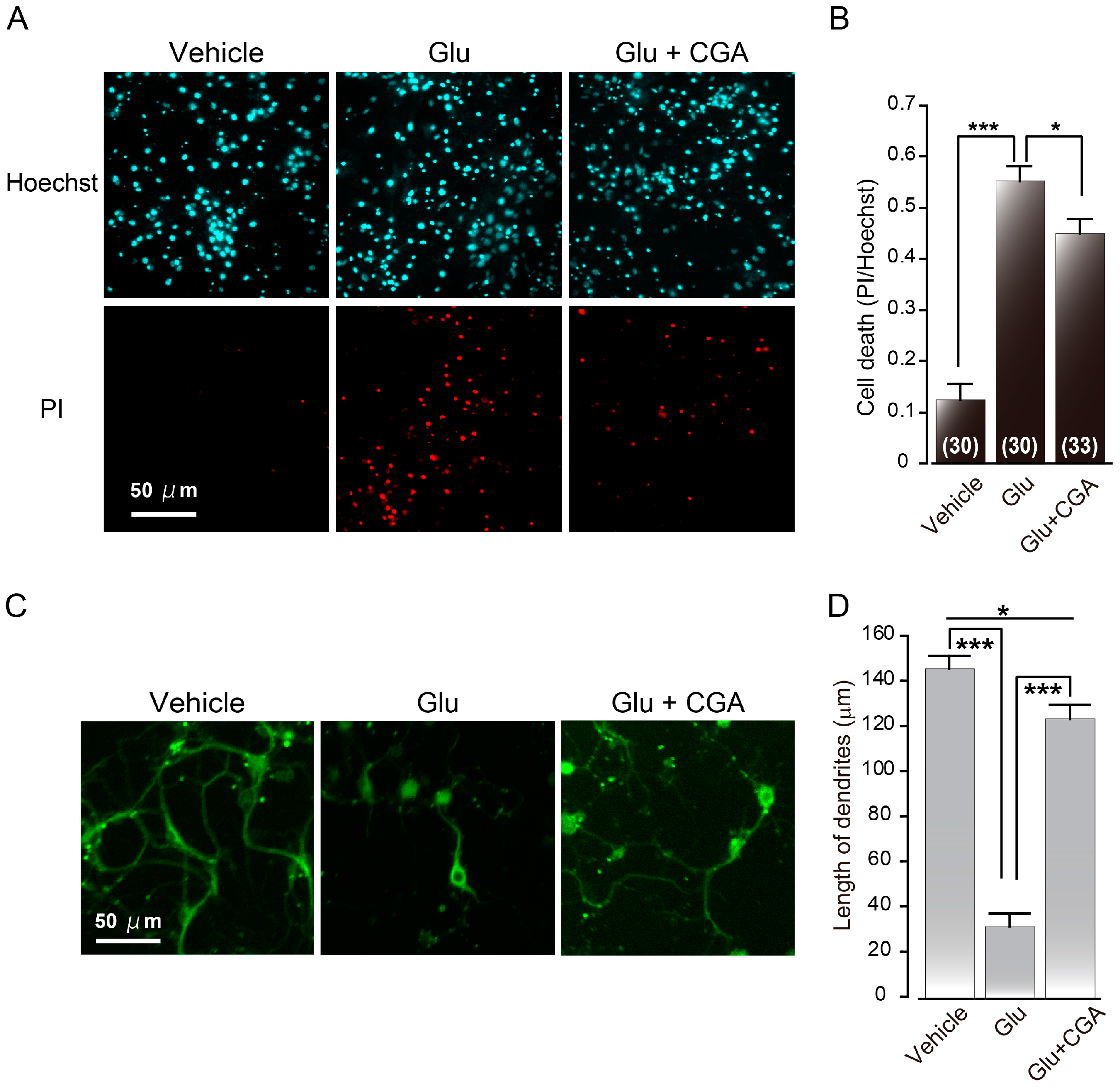
2.3. Chlorogenic Acid
3. Neuroprotection by H2S
3.1. H2S and Polysulfide
3.2. Neuroprotective Effect of H2S
4. Neuronal Cell Death by NO
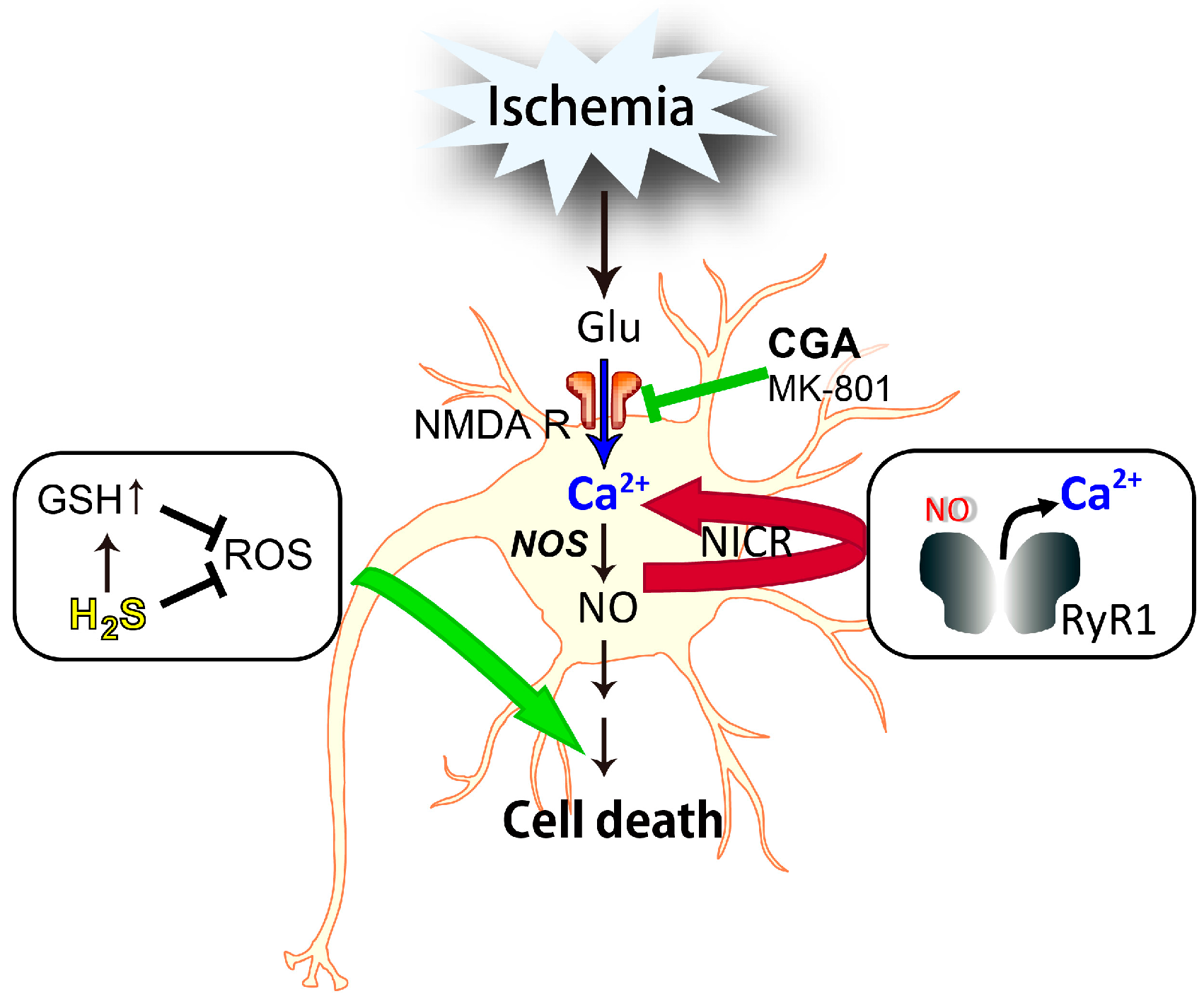
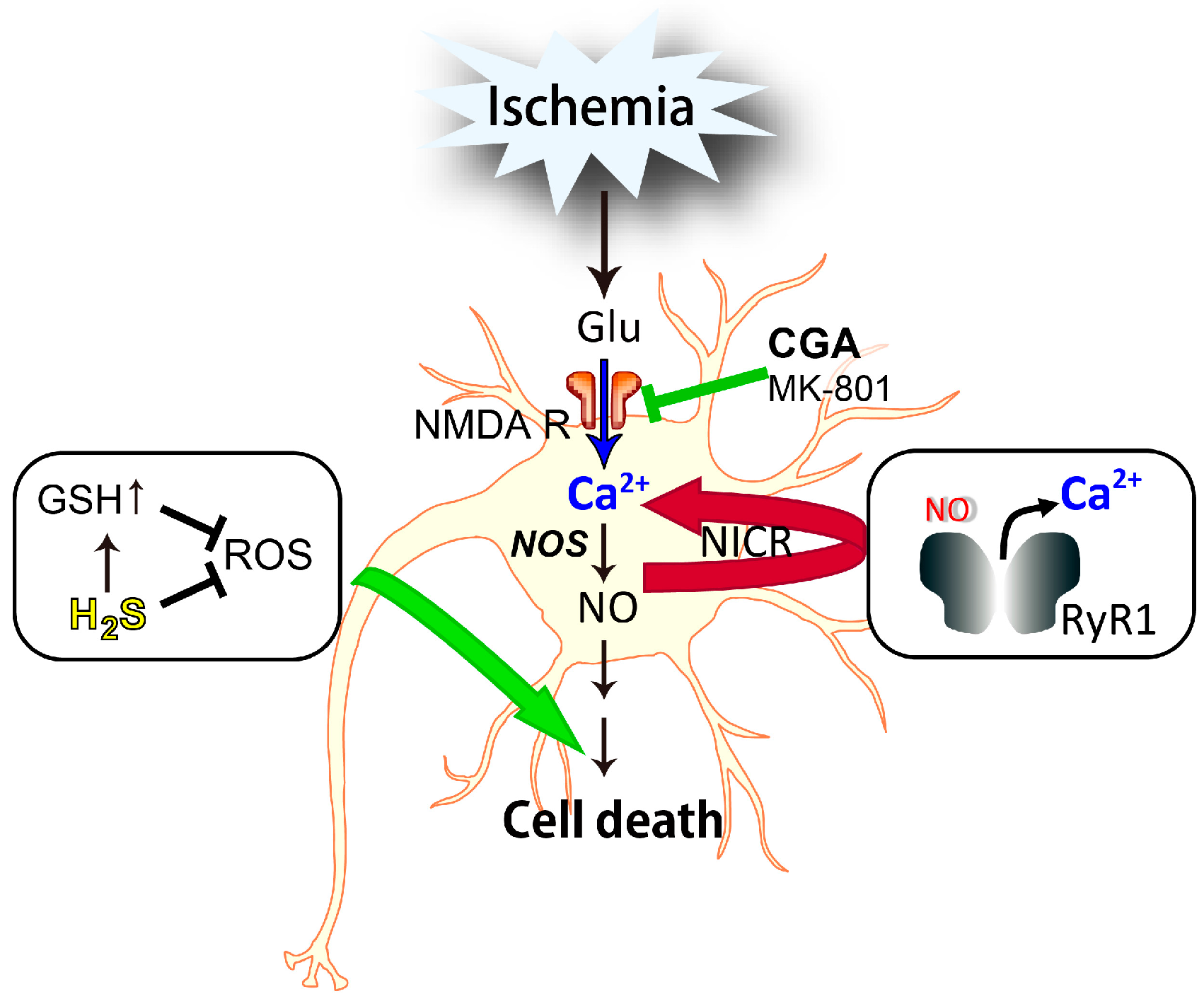
4.1. Nitric Oxide-Induced Calcium Release (NICR)
4.2. Involvement of NICR in NO-Induced Neuronal Cell Death
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 3MST | 3-Mercaptopyruvate sulfurtransferase |
| Aβ | β-amyloid |
| CA | Caffeic acid |
| CAPE | Caffeic acid phenethyl ester |
| CAT | Cysteine aminotransferase |
| CBS | Cystathionine β-synthase |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CGA | Chlorogenic acid |
| CICR | Calcium-induced calcium release |
| CSE | Cystathionine γ-lyase |
| DAO | d-amino acid oxidase |
| DHLA | Dihydrolipoic acid |
| eNOS | Endothelial nitric oxide synthase |
| ER | Endoplasmic reticulum |
| H2S | Hydrogen sulfide |
| iNOS | Inducible nitric oxide synthase |
| MCAO | Middle cerebral artery occlusion |
| MeHg | Methylmercury |
| NICR | Nitric oxide-induced calcium release |
| NMDA | N-methyl d-aspartate |
| nNOS | Neuronal nitric oxide synthase |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| Nrf2 | Nuclear factor erythroid-2 related factor 2 |
| PI | Propidium iodide |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
References
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Prentice, H.; Modi, J.P.; Wu, J.Y. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid. Med. Cell. Longev. 2015, 2015, 964518. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Sarkar, B.; Cholia, R.P.; Gautam, N.; Dhiman, M.; Mantha, A.K. APE1/Ref-1 as an emerging therapeutic target for various human diseases: Phytochemical modulation of its functions. Exp. Mol. Med. 2014, 46, e106. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical antioxidants as novel neuroprotective agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W.; Rothman, S.M. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Manucha, W. Mitochondrial dysfunction associated with nitric oxide pathways in glutamate neurotoxicity. Clin. Investig. Arterioscler. 2016. [Google Scholar] [CrossRef] [PubMed]
- Szydlowska, K.; Tymianski, M. Calcium, ischemia and excitotoxicity. Cell Calcium 2010, 47, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Anitha, M.; Nandhu, M.S.; Anju, T.R.; Jes, P.; Paulose, C.S. Targeting glutamate mediated excitotoxicity in Huntington’s disease: Neural progenitors and partial glutamate antagonist—Memantine. Med. Hypotheses 2011, 76, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Godinez-Rubi, M.; Rojas-Mayorquin, A.E.; Ortuno-Sahagun, D. Nitric oxide donors as neuroprotective agents after an ischemic stroke-related inflammatory reaction. Oxid. Med. Cell. Longev. 2013, 2013, 297357. [Google Scholar] [CrossRef] [PubMed]
- Kostandy, B.B. The role of glutamate in neuronal ischemic injury: The role of spark in fire. Neurol. Sci. 2012, 33, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Lipton, S.A. Cell death: Protein misfolding and neurodegenerative diseases. Apoptosis 2009, 14, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [PubMed]
- Bredt, D.S.; Snyder, S.H. Nitric oxide: A physiologic messenger molecule. Annu. Rev. Biochem. 1994, 63, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kawada, K.; Shiba, T.; Ogita, K. Endogenous nitric oxide generation linked to ryanodine receptors activates cyclic GMP/protein kinase G pathway for cell proliferation of neural stem/progenitor cells derived from embryonic hippocampus. J. Pharmacol. Sci. 2011, 115, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Lipton, S.A. Protein S-nitrosylation as a therapeutic target for neurodegenerative diseases. Trends Pharmacol. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614. [Google Scholar] [CrossRef] [PubMed]
- Asard, H. Ascorbate. In Redox Biochemistry; Banerjee, R., Ed.; John Wiley & Sons, Inc.: Hodoken, NJ, USA, 2007; pp. 22–27. [Google Scholar]
- Banhegyi, G.; Braun, L.; Csala, M.; Puskas, F.; Mandl, J. Ascorbate metabolism and its regulation in animals. Free Radic. Biol. Med. 1997, 23, 793–803. [Google Scholar] [CrossRef]
- Della Penna, D.; Pogson, B.J. Vitamin synthesis in plants: Tocopherols and carotenoids. Annu. Rev. Plant Biol. 2006, 57, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Li, A.N.; Li, S.; Zhang, Y.J.; Xu, X.R.; Chen, Y.M.; Li, H.B. Resources and biological activities of natural polyphenols. Nutrients 2014, 6, 6020–6047. [Google Scholar] [CrossRef] [PubMed]
- Brglez Mojzer, E.; Knez Hrncic, M.; Skerget, M.; Knez, Z.; Bren, U. Polyphenols: Extraction Methods, Antioxidative Action, Bioavailability and Anticarcinogenic Effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef] [PubMed]
- Barycki, J.J. Glutathione. In Redox Biochemicstry; Banerjee, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 11–22. [Google Scholar]
- Circu, M.L.; Aw, T.Y. Glutathione and modulation of cell apoptosis. Biochim. Biophys. Acta 2012, 1823, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Carlson, J.L.; Mody, V.C.; Cai, J.; Lynn, M.J.; Sternberg, P. Redox state of glutathione in human plasma. Free Radic. Biol. Med. 2000, 28, 625–635. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, T.; Yasuno, R.; Wada, H. Do mammalian cells synthesize lipoic acid? Identification of a mouse cDNA encoding a lipoic acid synthase located in mitochondria. FEBS Lett. 2001, 498, 16–21. [Google Scholar] [CrossRef]
- Reed, L.J.; Leach, F.R.; Koike, M. Studies on a lipoic acid-activating system. J. Biol. Chem. 1958, 232, 123–142. [Google Scholar] [PubMed]
- Smith, A.R.; Shenvi, S.V.; Widlansky, M.; Suh, J.H.; Hagen, T.M. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr. Med. Chem. 2004, 11, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [PubMed]
- Higdon, J.V.; Frei, B. Coffee and health: A review of recent human research. Crit. Rev. Food Sci. Nutr. 2006, 46, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.F.; Jacobs, D.R.J.; Carlsen, M.H.; Blomhoff, R. Consumption of coffee is associated with reduced risk of death attributed to inflammatory and cardiovascular diseases in the Iowa Women’s Health Study. Am. J. Clin. Nutr. 2006, 83, 1039–1046. [Google Scholar] [PubMed]
- Freedman, N.D.; Park, Y.; Abnet, C.C.; Hollenbeck, A.R.; Sinha, R. Association of coffee drinking with total and cause-specific mortality. N. Engl. J. Med. 2012, 366, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, J.H.; Bhatti, S.K.; Patil, H.R.; DiNicolantonio, J.J.; Lucan, S.C.; Lavie, C.J. Effects of habitual coffee consumption on cardiometabolic disease, cardiovascular health, and all-cause mortality. J. Am. Coll. Cardiol. 2013, 62, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Rebello, S.A.; van Dam, R.M. Coffee consumption and cardiovascular health: Getting to the heart of the matter. Curr. Cardiol. Rep. 2013, 15, 403. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, Y.; Iso, H.; Saito, I.; Yamagishi, K.; Yatsuya, H.; Ishihara, J.; Inoue, M.; Tsugane, S. The impact of green tea and coffee consumption on the reduced risk of stroke incidence in Japanese population: The Japan public health center-based study cohort. Stroke 2013, 44, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Virtamo, J.; Wolk, A. Coffee consumption and risk of stroke in women. Stroke 2011, 42, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, E.; Rodriguez-Artalejo, F.; Rexrode, K.M.; Logroscino, G.; Hu, F.B.; van Dam, R.M. Coffee consumption and risk of stroke in women. Circulation 2009, 119, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Croft, K.D. The chemistry and biological effects of flavonoids and phenolic acids. Ann. N. Y. Acad. Sci. 1998, 854, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Sul, D.; Kim, H.S.; Lee, D.; Joo, S.S.; Hwang, K.W.; Park, S.Y. Protective effect of caffeic acid against β-amyloid-induced neurotoxicity by the inhibition of calcium influx and tau phosphorylation. Life Sci. 2009, 84, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Oboh, G.; Agunloye, O.M.; Akinyemi, A.J.; Ademiluyi, A.O.; Adefegha, S.A. Comparative study on the inhibitory effect of caffeic and chlorogenic acids on key enzymes linked to Alzheimer’s disease and some pro-oxidant induced oxidative stress in rats’ brain-in vitro. Neurochem. Res. 2013, 38, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jin, M.; Pi, R.; Zhang, J.; Chen, M.; Ouyang, Y.; Liu, A.; Chao, X.; Liu, P.; Liu, J.; et al. Protective effects of caffeic acid and caffeic acid phenethyl ester against acrolein-induced neurotoxicity in HT22 mouse hippocampal cells. Neurosci. Lett. 2013, 535, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Scapagnini, G.; Vasto, S.; Abraham, N.G.; Caruso, C.; Zella, D.; Fabio, G. Modulation of Nrf2/ARE pathway by food polyphenols: A nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol. Neurobiol. 2011, 44, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Ma, Z.; Fontanilla, C.V.; Zhao, L.; Xu, Z.C.; Taggliabraci, V.; Johnstone, B.H.; Dodel, R.C.; Farlow, M.R.; Du, Y. Caffeic acid phenethyl ester prevents cerebellar granule neurons (CGNs) against glutamate-induced neurotoxicity. Neuroscience 2008, 155, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, N.A.; Martins, N.M.; Silva Rde, B.; Ferreira, R.S.; Sisti, F.M.; dos Santos, A.C. Caffeic acid phenethyl ester (CAPE) protects PC12 cells from MPP+ toxicity by inducing the expression of neuron-typical proteins. Neurotoxicology 2014, 45, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Wang, Q.; Choi, J.M.; Lee, S.; Cho, E.J. Protective role of caffeic acid in an Aβ25–35-induced-induced Alzheimer’s disease model. Nutr. Res. Pract. 2015, 9, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Barros Silva, R.; Santos, N.A.; Martins, N.M.; Ferreira, D.A.; Barbosa, F., Jr.; Oliveira Souza, V.C.; Kinoshita, A.; Baffa, O.; del-Bel, E.; Santos, A.C. Caffeic acid phenethyl ester protects against the dopaminergic neuronal loss induced by 6-hydroxydopamine in rats. Neuroscience 2013, 233, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Shi, B.; Luo, W.; Yang, J. The protective effect of caffeic acid on global cerebral ischemia-reperfusion injury in rats. Behav. Brain Funct. 2015, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Bättig, K.; Holmén, J.; Nehlig, A.; Zvartau, E.E. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol. Rev. 1999, 51, 83–133. [Google Scholar] [PubMed]
- Dunwiddie, T.V.; Masino, S.A. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 2001, 24, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Zhang, S.M.; Hernan, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, M.D.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Peterson, B.J.; Ahlskog, J.E.; Schaid, D.J.; Rocca, W.A. Smoking, alcohol, and coffee consumption preceding Parkinson’s disease: A case-control study. Neurology 2000, 55, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Clifford, M.N.; Knight, S.; Surucu, B.; Kuhnert, N. Characterization by LC-MSn of four new classes of chlorogenic acids in green coffee beans: Dimethoxycinnamoylquinic acids, diferuloylquinic acids, caffeoyl-dimethoxycinnamoylquinic acids, and feruloyl-dimethoxycinnamoylquinic acids. J. Agric. Food Chem. 2006, 54, 1957–1969. [Google Scholar] [CrossRef] [PubMed]
- Farah, A.; Monteiro, M.C.; Calado, V.; Franca, A.S.; Trugo, L.C. Correlation between cup quality and chemical attributes of Brazilian coffee. Food Chem. 2006, 98, 373–380. [Google Scholar] [CrossRef]
- Upadhyay, R.; Mohan Rao, L.J. An outlook on chlorogenic acids-occurrence, chemistry, technology, and biological activities. Crit. Rev. Food Sci. Nutr. 2013, 53, 968–984. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Lu, Y.; Bowman, L.L.; Qian, Y.; Castranova, V.; Ding, M. Inhibition of activator protein-1, NF-κB, and MAPKs and induction of phase 2 detoxifying enzyme activity by chlorogenic acid. J. Biol. Chem. 2005, 280, 27888–27895. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shi, W.; Li, Y.; Zhou, Y.; Hu, X.; Song, C.; Ma, H.; Wang, C.; Li, Y. Neuroprotective effects of chlorogenic acid against apoptosis of PC12 cells induced by methylmercury. Environ. Toxicol. Pharmacol. 2008, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.J.; Yen, G.C. Chemopreventive effects of dietary phytochemicals against cancer invasion and metastasis: Phenolic acids, monophenol, polyphenol, and their derivatives. Cancer Treat. Rev. 2012, 38, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.Y.; Cosma, G.; Gardner, H.; Castranova, V.; Vallyathan, V. Effect of chlorogenic acid on hydroxyl radical. Mol. Cell. Biochem. 2003, 247, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, J.S.; Jang, H.J.; Kim, S.M.; Chang, M.S.; Park, S.H.; Kim, K.S.; Bae, J.; Park, J.W.; Lee, B.; et al. Chlorogenic acid ameliorates brain damage and edema by inhibiting matrix metalloproteinase-2 and 9 in a rat model of focal cerebral ischemia. Eur. J. Pharmacol. 2012, 689, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Jang, Y.J.; Hwang, M.K.; Kang, N.J.; Lee, K.W.; Lee, H.J. Attenuation of oxidative neuronal cell death by coffee phenolic phytochemicals. Mutat. Res. 2009, 661, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Pavlica, S.; Gebhardt, R. Protective effects of ellagic and chlorogenic acids against oxidative stress in PC12 cells. Free Radic. Res. 2005, 39, 1377–1390. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.F.; LeBel, C.P.; Bondy, S.C. Reactive oxygen species formation as a biomarker of methylmercury and trimethyltin neurotoxicity. Neurotoxicology 1992, 13, 637–648. [Google Scholar] [PubMed]
- Sarafian, T.A.; Vartavarian, L.; Kane, D.J.; Bredesen, D.E.; Verity, M.A. Bcl-2 expression decreases methyl mercury-induced free-radical generation and cell killing in a neural cell line. Toxicol. Lett. 1994, 74, 149–155. [Google Scholar] [CrossRef]
- Ito, H.; Sun, X.L.; Watanabe, M.; Okamoto, M.; Hatano, T. Chlorogenic acid and its metabolite m-coumaric acid evoke neurite outgrowth in hippocampal neuronal cells. Biosci. Biotechnol. Biochem. 2008, 72, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Yamazawa, T. Chlorogenic acid, a polyphenol in coffee, protects neurons against glutamate neurotoxicity. Life Sci. 2015, 139, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Kubota, J.; Sekiya, H.; Hirose, K.; Okubo, Y.; Iino, M. Calcium-dependent N-cadherin up-regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc. Natl. Acad. Sci. USA 2013, 110, 11612–11617. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Yamazawa, T.; Okubo, Y.; Iino, M. Temporal switching and cell-to-cell variability in Ca2+ release activity in mammalian cells. Mol. Syst. Biol. 2009, 5, 247. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, L.R.; Francom, D.; Dieken, F.P.; Taylor, J.D.; Warenycia, M.W.; Reiffenstein, R.J.; Dowling, G. Determination of sulfide in brain tissue by gas dialysis/ion chromatography: Postmortem studies and two case reports. J. Anal. Toxicol. 1989, 13, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; Gould, D.H. Determination of sulfide in brain tissue and rumen fluid by ion-interaction reversed-phase high-performance liquid chromatography. J. Chromatogr. 1990, 526, 540–545. [Google Scholar] [CrossRef]
- Warenycia, M.W.; Goodwin, L.R.; Benishin, C.G.; Reiffenstein, R.J.; Francom, D.M.; Taylor, J.D.; Dieken, F.P. Acute hydrogen sulfide poisoning. Demonstration of selective uptake of sulfide by the brainstem by measurement of brain sulfide levels. Biochem. Pharmacol. 1989, 38, 973–981. [Google Scholar] [CrossRef]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Shibuya, N.; Kimura, Y.; Nagahara, N.; Ogasawara, Y.; Kimura, H. Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 2011, 439, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from d-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Mikami, Y.; Osumi, K.; Tsugane, M.; Oka, J.; Kimura, H. Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. 2013, 27, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Toyofuku, Y.; Koike, S.; Shibuya, N.; Nagahara, N.; Lefer, D.; Ogasawara, Y.; Kimura, H. Identification of H2S3 and H2S produced by 3-mercaptopyruvate sulfurtransferase in the brain. Sci. Rep. 2015, 5, 14774. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Tsugane, M.; Oka, J.; Kimura, H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004, 18, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Mikami, Y.; Kimura, Y.; Nagahara, N.; Kimura, H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 2009, 146, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [PubMed]
- Ishii, I.; Akahoshi, N.; Yamada, H.; Nakano, S.; Izumi, T.; Suematsu, M. Cystathionine γ-lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem. 2010, 285, 26358–26368. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite “scavenger”? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Cheung, N.S.; Zhu, Y.Z.; Chu, S.H.; Siau, J.L.; Wong, B.S.; Armstrong, J.S.; Moore, P.K. Hydrogen sulphide: A novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem. Biophys. Res. Commun. 2005, 326, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Coetzee, W.A.; Lefer, D.J. Novel insights into hydrogen sulfide-mediated cytoprotection. Antioxid. Redox Signal. 2010, 12, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Ogasawara, Y.; Shibuya, N.; Kimura, H.; Ishii, K. Polysulfide exerts a protective effect against cytotoxicity caused by t-buthylhydroperoxide through Nrf2 signaling in neuroblastoma cells. FEBS Lett. 2013, 587, 3548–3555. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Dargusch, R.; Schubert, D.; Kimura, H. Hydrogen sulfide protects HT22 neuronal cells from oxidative stress. Antioxid. Redox Signal. 2006, 8, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Johansen, D.; Ytrehus, K.; Baxter, G.F. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury-Evidence for a role of K ATP channels. Basic Res. Cardiol. 2006, 101, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Shibuya, N.; Kimura, Y.; Nagahara, N.; Yamada, M.; Kimura, H. Hydrogen sulfide protects the retina from light-induced degeneration by the modulation of Ca2+ influx. J. Biol. Chem. 2011, 286, 39379–39386. [Google Scholar] [CrossRef] [PubMed]
- Noell, W.K.; Walker, V.S.; Kang, B.S.; Berman, S. Retinal damage by light in rats. Investig. Ophthalmol. 1966, 5, 450–473. [Google Scholar]
- Wenzel, A.; Grimm, C.; Samardzija, M.; Reme, C.E. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog. Retin. Eye Res. 2005, 24, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, S.M.; Cinel, I. Bench-to-bedside review: Nitric oxide in critical illness-update 2008. Crit. Care 2009, 13, 218. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.; Sterk, P.J.; Gaston, B.; Folkerts, G. Nitric oxide in health and disease of the respiratory system. Physiol. Rev. 2004, 84, 731–765. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Koesling, D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ. Res. 2003, 93, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Shahani, N.; Sawa, A. Nitric oxide signaling and nitrosative stress in neurons: Role for S-nitrosylation. Antioxid. Redox Signal. 2011, 14, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Jaffrey, S.R.; Erdjument-Bromage, H.; Ferris, C.D.; Tempst, P.; Snyder, S.H. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001, 3, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Eu, J.P.; Sun, J.; Xu, L.; Stamler, J.S.; Meissner, G. The skeletal muscle calcium release channel: Coupled O2 sensor and NO signaling functions. Cell 2000, 102, 499–509. [Google Scholar] [CrossRef]
- Endo, M. Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 2009, 89, 1153–1176. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, S.; Yamazawa, T.; Chen, Y.; Ito, A.; Murayama, T.; Oyamada, H.; Kurebayashi, N.; Sato, O.; Watanabe, M.; Mori, N.; et al. Nitric oxide-induced calcium release via ryanodine receptors regulates neuronal function. EMBO J. 2012, 31, 417–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakizawa, S.; Yamazawa, T.; Iino, M. Nitric oxide-induced calcium release: Activation of type 1 ryanodine receptor by endogenous nitric oxide. Channels (Austin) 2013, 7, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadecola, C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997, 20, 132–139. [Google Scholar] [CrossRef]
- Malinski, T.; Bailey, F.; Zhang, Z.G.; Chopp, M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 1993, 13, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, P.L.; Panahian, N.; Dalkara, T.; Fishman, M.C.; Moskowitz, M.A. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science 1994, 265, 1883–1885. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Kanemaru, K.; Okubo, Y.; Nakaune, T.; Suzuki, J.; Shibata, K.; Sugiyama, H.; Koyama, R.; Murayama, T.; Ito, A.; et al. Nitric oxide-induced activation of the type 1 ryanodine receptor receptor is critical for epileptic seizure-induced neuronal cell death. EBioMedicine 2016, 11, 253–261. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikami, Y.; Kakizawa, S.; Yamazawa, T. Essential Roles of Natural Products and Gaseous Mediators on Neuronal Cell Death or Survival. Int. J. Mol. Sci. 2016, 17, 1652. https://doi.org/10.3390/ijms17101652
Mikami Y, Kakizawa S, Yamazawa T. Essential Roles of Natural Products and Gaseous Mediators on Neuronal Cell Death or Survival. International Journal of Molecular Sciences. 2016; 17(10):1652. https://doi.org/10.3390/ijms17101652
Chicago/Turabian StyleMikami, Yoshinori, Sho Kakizawa, and Toshiko Yamazawa. 2016. "Essential Roles of Natural Products and Gaseous Mediators on Neuronal Cell Death or Survival" International Journal of Molecular Sciences 17, no. 10: 1652. https://doi.org/10.3390/ijms17101652