
Assessment of Amino Acid/Drug Transporters for Renal Transport of [18F]Fluciclovine (anti-[18F]FACBC) in Vitro

 ,
,
Abstract
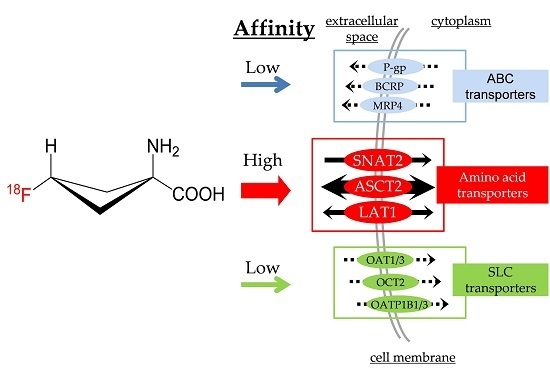
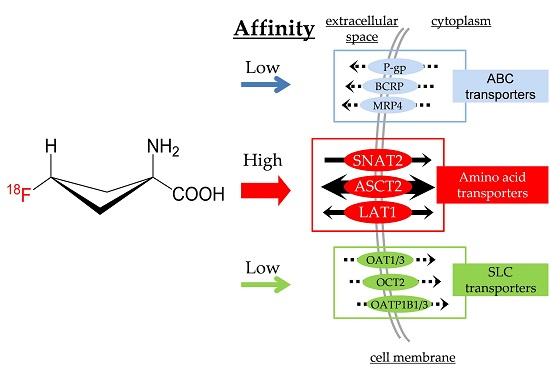
:
1. Introduction
2. Results
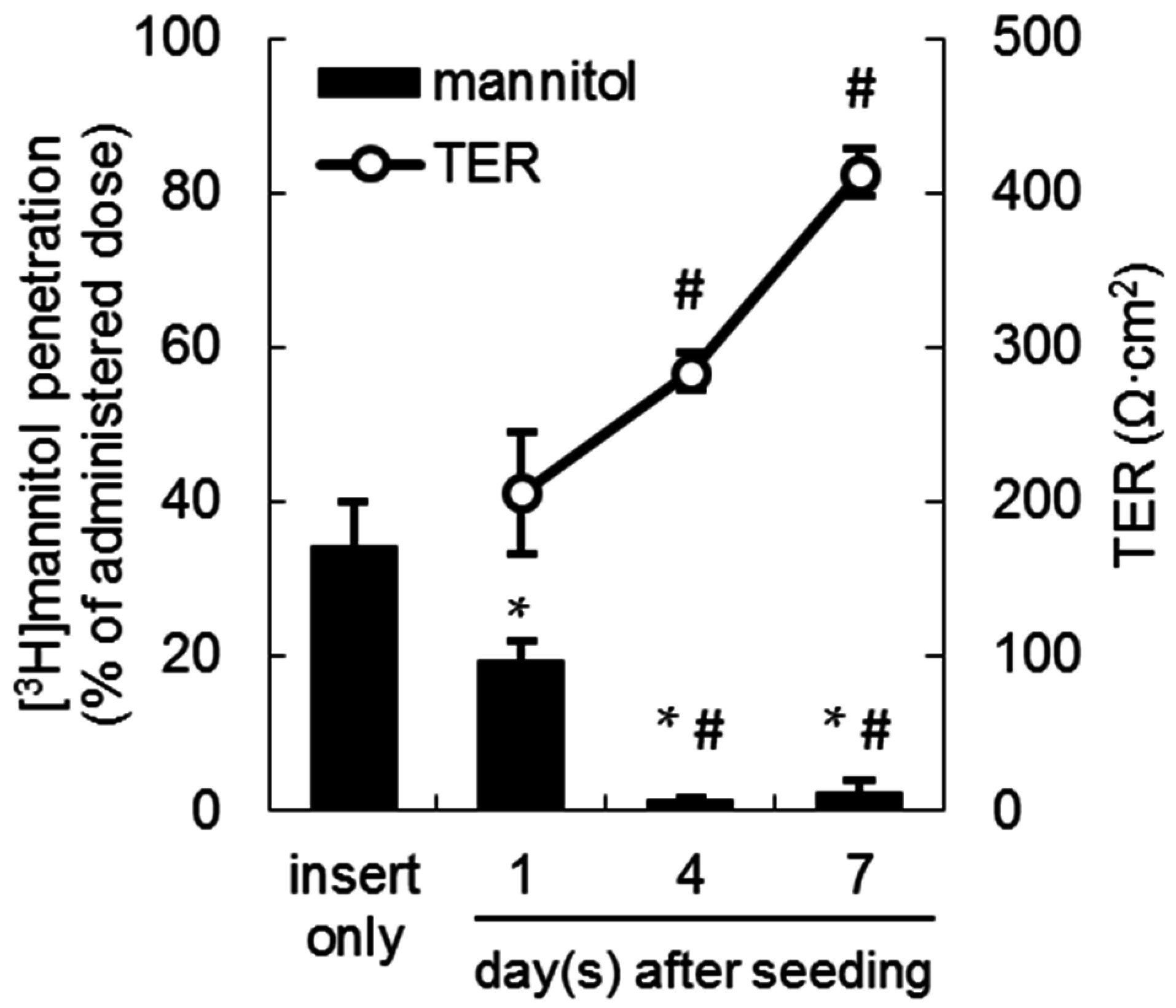
2.1. Tight Junction Formation in the LLC-PK1 Monolayer Model
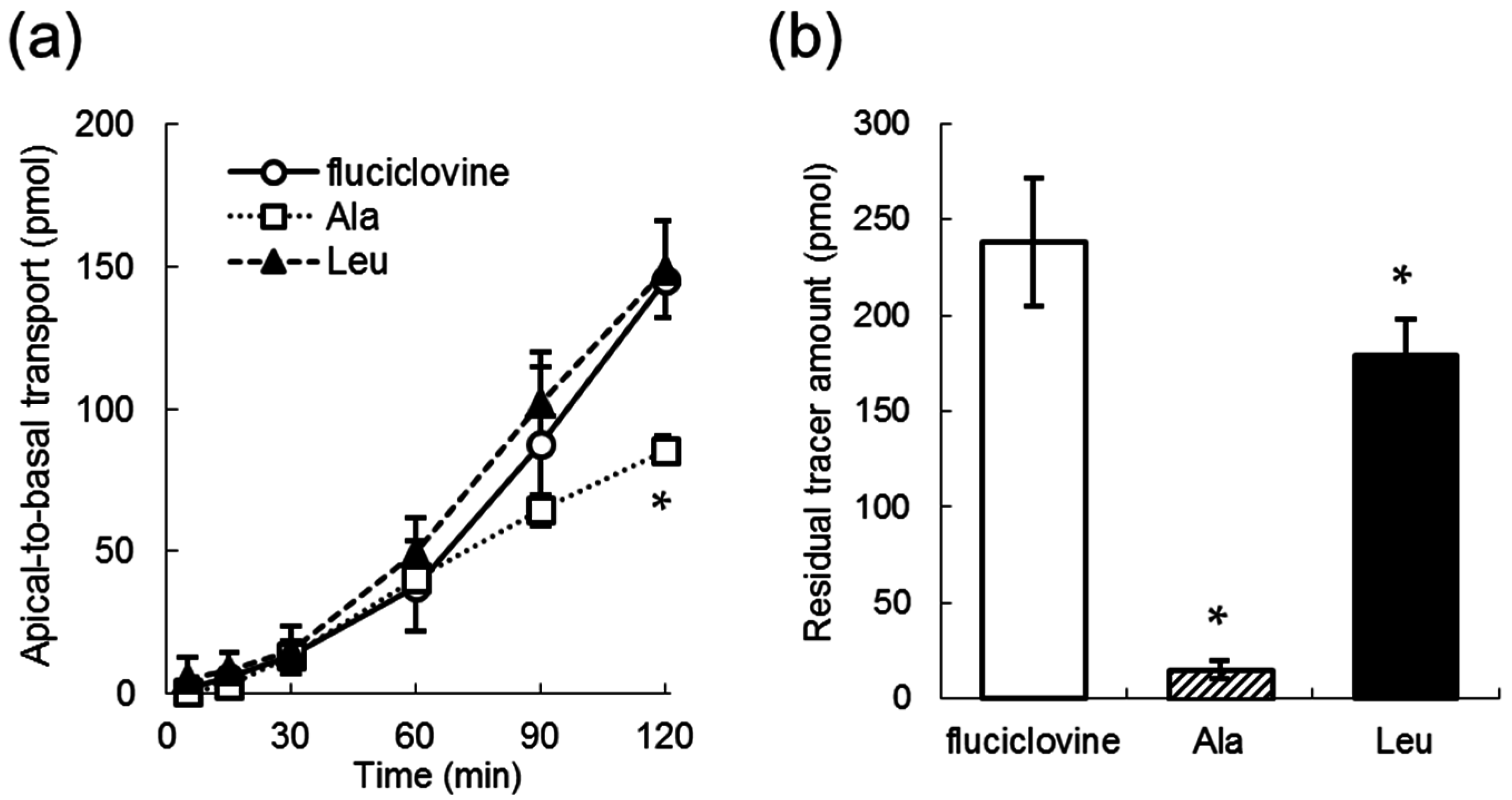
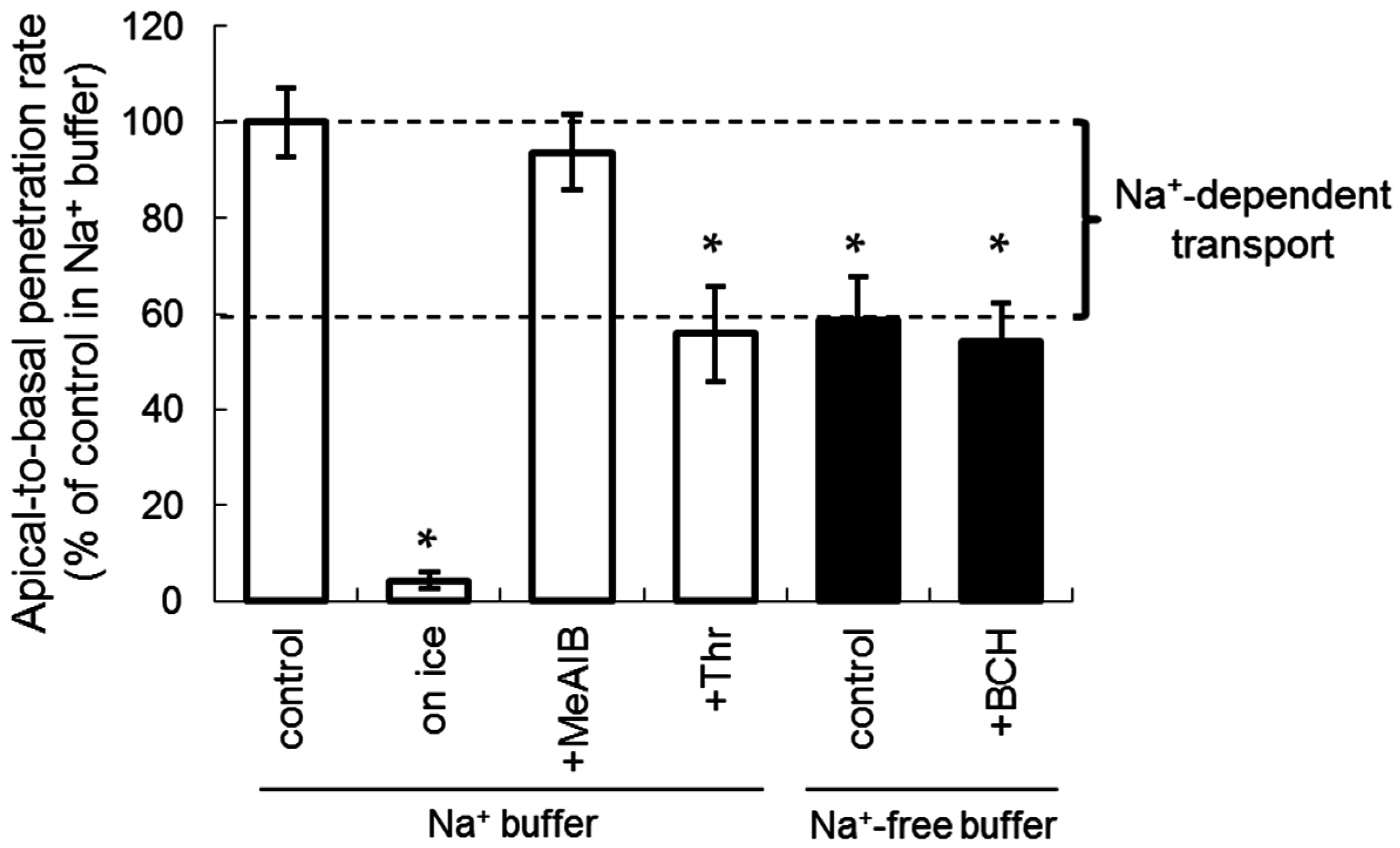
2.2. Apical-to-Basal Transport of Amino Acid Tracers through the LLC-PK1 Monolayers
2.3. The Inhibition of Apical-to-Basal Transport in the LLC-PK1 Monolayers by Amino Acids
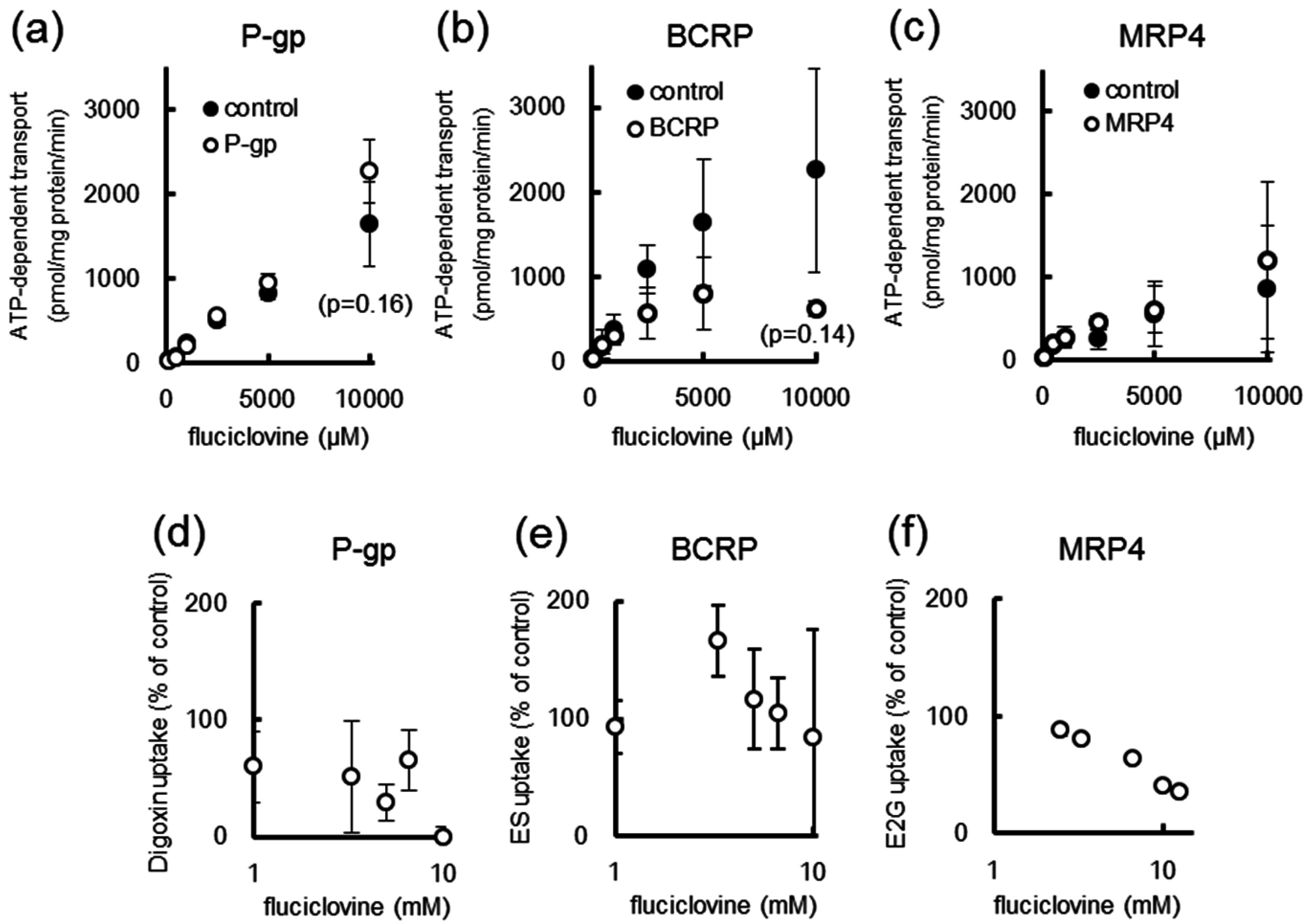
2.4. [14C]Fluciclovine Transport via ATP-binding Cassette (ABC) Transporters and the Inhibitory Effect of Fluciclovine on ABC Transporters
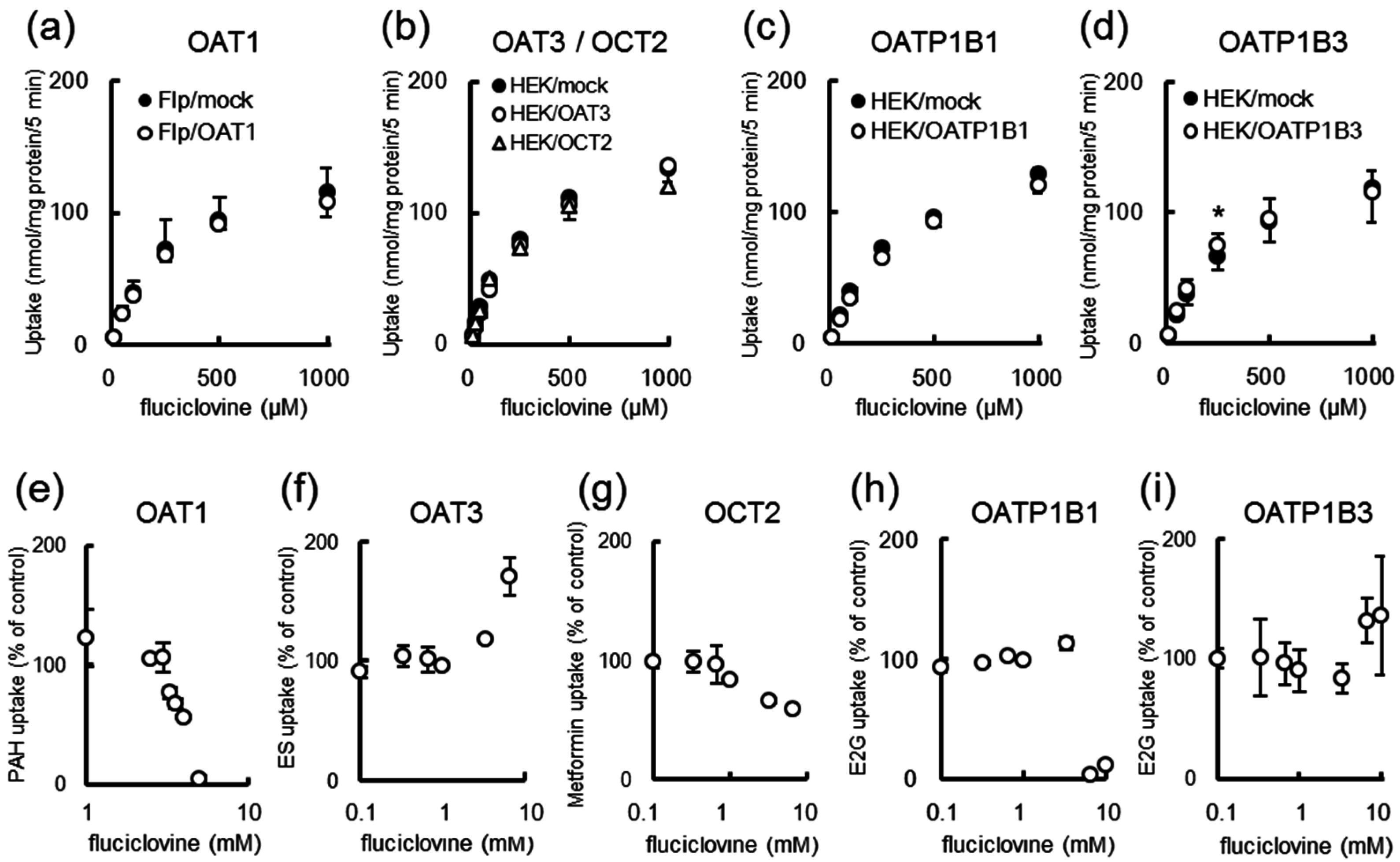
2.5. [14C]Fluciclovine Transport via Solute Carrier (SLC) Transporters and the Inhibition of SLC Transporters by Fluciclovine
3. Discussion
4. Materials and Methods
4.1. Reagents and Radioisotope-Labeled Tracers
4.2. Monolayer Establishment Using LLC-PK1 Cells
4.3. Apical-to-Basal Transport Using LLC-PK1 Monolayers
4.4. Inhibition Study Using the LLC-PK1 Monolayer Model
4.5. Uptake and Inhibitory Assay Using ABC Transporter-Expressing Vesicles
4.6. Uptake and Inhibitory Assay Using SLC Transporter-Expressing Cells
4.7. Data and Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- The US Food and Drug Administration. Drug Trails Snapshots: Axumin. Available online: http://www.fda.gov/drugs/informationondrugs/ucm505750.htm (accessed on 13 September 2016).
- Kondo, A.; Ishii, H.; Aoki, S.; Suzuki, M.; Nagasawa, H.; Kubota, K.; Minamimoto, R.; Arakawa, A.; Tominaga, M.; Arai, H. Phase IIa clinical study of [18F]fluciclovine: Efficacy and safety of a new PET tracer for brain tumors. Ann. Nucl. Med. 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Inoue, Y.; Fujimoto, H.; Yonese, J.; Tanabe, K.; Fukasawa, S.; Inoue, T.; Saito, S.; Ueno, M.; Otaka, A. Diagnostic performance and safety of NMK36 (trans-1-amino-3-[18F]fluorocyclobutanecarboxylic acid)-PET/CT in primary prostate cancer: Multicenter Phase IIb clinical trial. Jpn. J. Clin. Oncol. 2016, 46, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Boellaard, R.; Delgado-Bolton, R.; Oyen, W.J.; Giammarile, F.; Tatsch, K.; Eschner, W.; Verzijlbergen, F.J.; Barrington, S.F.; Pike, L.C.; Weber, W.A.; et al. European Association of Nuclear Medicine. FDG PET/CT: EANM procedure guidelines for tumour imaging: Version 2.0. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 328–354. [Google Scholar] [CrossRef] [PubMed]
- Pauleit, D.; Floeth, F.; Herzog, H.; Hamacher, K.; Tellmann, L.; Müller, H.W.; Coenen, H.H.; Langen, K.J. Whole-body distribution and dosimetry of O-(2-[18F]fluoroethyl)-l-tyrosine. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Inoue, Y.; Ikeda, Y.; Kikuchi, K.; Hara, T.; Taguchi, C.; Tokushige, T.; Maruo, H.; Takeda, T.; Nakamura, T.; et al. Phase I clinical study of NMK36: A new PET tracer with the synthetic amino acid analogue anti-[18F]FACBC. Ann. Nucl. Med. 2011, 25, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.M.; Nanni, C.; Fanti, S.; Oka, S.; Okudaira, H.; Inoue, Y.; Sorensen, J.; Owenius, R.; Choyke, P.; Turkbey, B.; et al. Anti-1-amino-3-[18F]-fluorocyclobutane-1-carboxylic acid: Physiologic uptake patterns, incidental findings, and variants that may simulate disease. J. Nucl. Med. 2014, 55, 1986–1992. [Google Scholar] [CrossRef] [PubMed]
- Okudaira, H.; Shikano, N.; Nishii, R.; Miyagi, T.; Yoshimoto, M.; Kobayashi, M.; Ohe, K.; Nakanishi, T.; Tamai, I.; Namiki, M.; et al. Putative transport mechanism and intracellular fate of trans-1-amino-3-[18F]-fluorocyclobutanecarboxylic acid in human prostate cancer. J. Nucl. Med. 2011, 52, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Okudaira, H.; Yoshida, Y.; Schuster, D.M.; Goodman, M.M.; Shirakami, Y. Transport mechanisms of trans-1-amino-3-fluoro[1-14C]cyclobutanecarboxylic acid in prostate cancer cells. Nucl. Med. Biol. 2012, 39, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Oka, S.; Okudaira, H.; Schuster, D.M.; Goodman, M.M.; Kawai, K.; Shirakami, Y. Comparative evaluation of transport mechanisms of trans-1-amino-3-[18F]fluorocyclobutanecarboxylic acid and l-[methyl-11C]methionine in human glioma cell lines. Brain Res. 2013, 1535, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Okudaira, H.; Nakanishi, T.; Oka, S.; Kobayashi, M.; Tamagami, H.; Schuster, D.M.; Goodman, M.M.; Shirakami, Y.; Tamai, I.; Kawai, K. Kinetic analyses of trans-1-amino-3-[18F]fluorocyclobutanecarboxylic acid transport in Xenopus laevis oocytes expressing human ASCT2 and SNAT2. Nucl. Med. Biol. 2013, 40, 670–675. [Google Scholar] [CrossRef] [PubMed]
- BioParadigms. SLC Tables. Available online: http://slc.bioparadigms.org/ (accessed on 13 September 2016).
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Kanai, Y.; Kim, D.K.; Wempe, M.F.; Chairoungdua, A.; Morimoto, E.; Anders, M.W.; Endou, H. Transport of amino acid-related compounds mediated by l-type amino acid transporter 1 (LAT1): Insights into the mechanisms of substrate recognition. Mol. Pharmacol. 2002, 61, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Dickens, D.; Webb, S.D.; Antonyuk, S.; Giannoudis, A.; Owen, A.; Radisch, S.; Hasnain, S.S.; Pirmohamed, M. Transport of gabapentin by LAT1 (SLC7A5). Biochem. Pharmacol. 2013, 85, 1672–1683. [Google Scholar] [CrossRef] [PubMed]
- Kuhne, A.; Tzvetkov, M.V.; Hagos, Y.; Lage, H.; Burckhardt, G.; Brockmoller, J. Influx and efflux transport as determinants of melphalan cytotoxicity: Resistance to melphalan in MDR1 overexpressing tumor cell lines. Biochem. Pharmacol. 2009, 78, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Urban, T.J.; Brown, C.; Castro, R.A.; Shah, N.; Mercer, R.; Huang, Y.; Brett, C.M.; Burchard, E.G.; Giacomini, K.M. Effects of genetic variation in the novel organic cation transporter, OCTN1, on the renal clearance of gabapentin. Clin. Pharmacol. Ther. 2008, 83, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Bader, P.; Schilling, F.; Schlaud, M.; Girgert, R.; Handgretinger, R.; Klingebiel, T.; Treuner, J.; Liu, C.; Niethammer, D.; Beck, J.F. Expression analysis of multidrug resistance associated genes in neuroblastomas. Oncol. Rep. 1999, 6, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Giesing, M.; Driesel, G.; Molitor, D.; Suchy, B. Molecular phenotyping of circulating tumour cells in patients with prostate cancer: Prediction of distant metastases. BJU Int. 2012, 110, E1202–E1211. [Google Scholar] [CrossRef] [PubMed]
- LeVier, D.G.; McCoy, D.E.; Spielman, W.S. Functional localization of adenosine receptor-mediated pathways in the LLC-PK1 renal cell line. Am. J. Physiol. 1992, 263, C729–C735. [Google Scholar] [PubMed]
- Soares-da-Silva, P.; Serrão, P.; Fraga, S.; Pinho, M.J. Expression and function of LAT1, a neutral amino acid exchanger, in renal porcine epithelial cell line LLC-PK. Acta Physiol. Scand. 2005, 185, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol. Rev. 2008, 88, 249–286. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas Project. The Human Protein Atlas: SLC38A2. Available online: http://www.proteinatlas.org/ENSG00000134294-SLC38A2/tissue (accessed on 13 September 2016).
- Suvannasankha, A.; Minderman, H.; O’Loughlin, K.L.; Nakanishi, T.; Ford, L.A.; Greco, W.R.; Wetzler, M.; Ross, D.D.; Baer, M.R. Breast cancer resistance protein (BCRP/MXR/ABCG2) in adult acute lymphoblastic leukaemia: Frequent expression and possible correlation with shorter disease-free survival. Br. J. Haematol. 2004, 127, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Nye, J.A.; Schuster, D.M.; Yu, W.; Camp, V.M.; Goodman, M.M.; Votaw, J.R. Biodistribution and radiation dosimetry of the synthetic nonmetabolized amino acid analogue anti-[18F]-FACBC in humans. J. Nucl. Med. 2007, 48, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, T.; Beattie, B.; Gogiberidze, G.; Montiel, J.; Cai, S.; Lassman, A.; Schoder, H.; Pillarsetty, N.; Goodman, M.; Blasberg, R. [18F]FACBC Imaging of recurrent gliomas: A comparison with [11C]methionine and MRI. J. Nucl. Med. 2006, 47, 79. [Google Scholar]
- O'Kane, R.L.; Vina, J.R.; Simpson, I.; Hawkins, R.A. Na+-dependent neutral amino acid transporters A, ASC, and N of the blood-brain barrier: Mechanisms for neutral amino acid removal. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E622–E629. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.M.; Davis, J.C.; Snyder, S.E.; Butch, E.R.; Vavere, A.L.; Kocak, M.; Shulkin, B.L. Evaluation of the biodistribution of [11C]-methionine in children and young adults. J. Nucl. Med. 2013, 54, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Trencsenyi, G.; Marian, T.; Lajtos, I.; Krasznai, Z.; Balkay, L.; Emri, M.; Mikecz, P.; Goda, K.; Szaloki, G.; Juhasz, I.; et al. [18F]DG, [18F]FLT, [18F]FAZA, and [11C]-methionine are suitable tracers for the diagnosis and in vivo follow-up of the efficacy of chemotherapy by miniPET in both multidrug resistant and sensitive human gynecologic tumor xenografts. BioMed Res. Int. 2014, 2014, 787365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y.; Huang, S.M. Scientific and regulatory perspectives on metabolizing enzyme-transporter interplay and its role in drug interactions: Challenges in predicting drug interactions. Mol. Pharm. 2009, 6, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Stout, D.B. Chapter 2 Imaging in Drug Development: Animal models, handling and physiological constraints. In Pharmaco-Imaging in Drug and Biologics Development Fundamentals and Applications; Moyer, B.R., Cheruvu, N.P.S., Hu, T., Eds.; Springer: New York, NY, USA, 2014; pp. 45–62. [Google Scholar]
- Tournier, N.; Saba, W.; Goutal, S.; Gervais, P.; Valette, H.; Scherrmann, J.M.; Bottlaender, M.; Cisternino, S. Influence of P-Glycoprotein inhibition or deficiency at the blood-brain barrier on [18F]-2-fluoro-2-deoxy-d-glucose ([18F]-FDG) Brain Kinetics. AAPS J. 2015, 17, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Isohashi, K.; Shimosegawa, E.; Kato, H.; Kanai, Y.; Naka, S.; Fujino, K.; Watabe, H.; Hatazawa, J. Optimization of [11C]methionine PET study: Appropriate scan timing and effect of plasma amino acid concentrations on the SUV. EJNMMI Res. 2013, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Hatazawa, J.; Murakami, M.; Miura, S.; Iida, H.; Bloomfield, P.M.; Kanno, I.; Fukuda, H.; Uemura, K. Aging effect on neutral amino acid transport at the blood-brain barrier measured with l-[2-18F]-fluorophenylalanine and PET. J. Nucl. Med. 1995, 36, 1232–1237. [Google Scholar] [PubMed]
- Nishioka, M.; Imaizumi, A.; Ando, T.; Tochikubo, O. The overnight effect of dietary energy balance on postprandial plasma free amino acid (PFAA) profiles in Japanese adult men. PLoS ONE 2013, 8, e6292913. [Google Scholar] [CrossRef] [PubMed]
- Sasajima, T.; Ono, T.; Shimada, N.; Doi, Y.; Oka, S.; Kanagawa, M.; Baden, A.; Mizoi, K. Trans-1-amino-3-[18F]-fluorocyclobutanecarboxylic acid (anti-[18F]-FACBC) is a feasible alternative to [11C]-methyl-l-methionine and magnetic resonance imaging for monitoring treatment response in gliomas. Nucl. Med. Biol. 2013, 40, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Nakakariya, M.; Shima, Y.; Shirasaka, Y.; Mitsuoka, K.; Nakanishi, T.; Tamai, I. Organic anion transporter OAT1 is involved in renal handling of citrulline. Am. J. Physiol. Renal Physiol. 2009, 297, F71–F79. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Nakanishi, T.; Nishi, K.; Higaki, Y.; Okudaira, H.; Ono, M.; Tsujiuchi, T.; Mizutani, A.; Nishii, R.; Tamai, I.; et al. Transport mechanisms of hepatic uptake and bile excretion in clinical hepatobiliary scintigraphy with 99mTc-N-pyridoxyl-5-methyltryptophan. Nucl. Med. Biol. 2014, 41, 338–342. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporters | Km (mM) | IC50 (mM) |
|---|---|---|
| Efflux Transporters | ||
| P-gp | N.D. | 2.95 ± 2.56 |
| BCRP 1 | 1.34 ± 1.45 | N.D. |
| MRP4 1 | 15.3 ± 44.1 | 9.28 ± 1.90 |
| Uptake transporters | ||
| OAT1 2 | 258 ± 48.2 | 3.98 ± 1.77 |
| OAT3 3 | N.D. | |
| OCT2 3 | 275 ± 36.6 | 8.95 ± 1.96 |
| OATP1B1 4 | 401 ± 41.0 | 5.41 ± 2.49 |
| OATP1B3 5 | 238 ± 82.5 | N.D. |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ono, M.; Baden, A.; Okudaira, H.; Kobayashi, M.; Kawai, K.; Oka, S.; Yoshimura, H. Assessment of Amino Acid/Drug Transporters for Renal Transport of [18F]Fluciclovine (anti-[18F]FACBC) in Vitro. Int. J. Mol. Sci. 2016, 17, 1730. https://doi.org/10.3390/ijms17101730
Ono M, Baden A, Okudaira H, Kobayashi M, Kawai K, Oka S, Yoshimura H. Assessment of Amino Acid/Drug Transporters for Renal Transport of [18F]Fluciclovine (anti-[18F]FACBC) in Vitro. International Journal of Molecular Sciences. 2016; 17(10):1730. https://doi.org/10.3390/ijms17101730
Chicago/Turabian StyleOno, Masahiro, Atsumi Baden, Hiroyuki Okudaira, Masato Kobayashi, Keiichi Kawai, Shuntaro Oka, and Hirokatsu Yoshimura. 2016. "Assessment of Amino Acid/Drug Transporters for Renal Transport of [18F]Fluciclovine (anti-[18F]FACBC) in Vitro" International Journal of Molecular Sciences 17, no. 10: 1730. https://doi.org/10.3390/ijms17101730