
Neuropeptide Y1 Receptor Regulates Glucocorticoid-Induced Inhibition of Osteoblast Differentiation in Murine MC3T3-E1 Cells via ERK Signaling

Abstract
:1. Introduction
2. Results
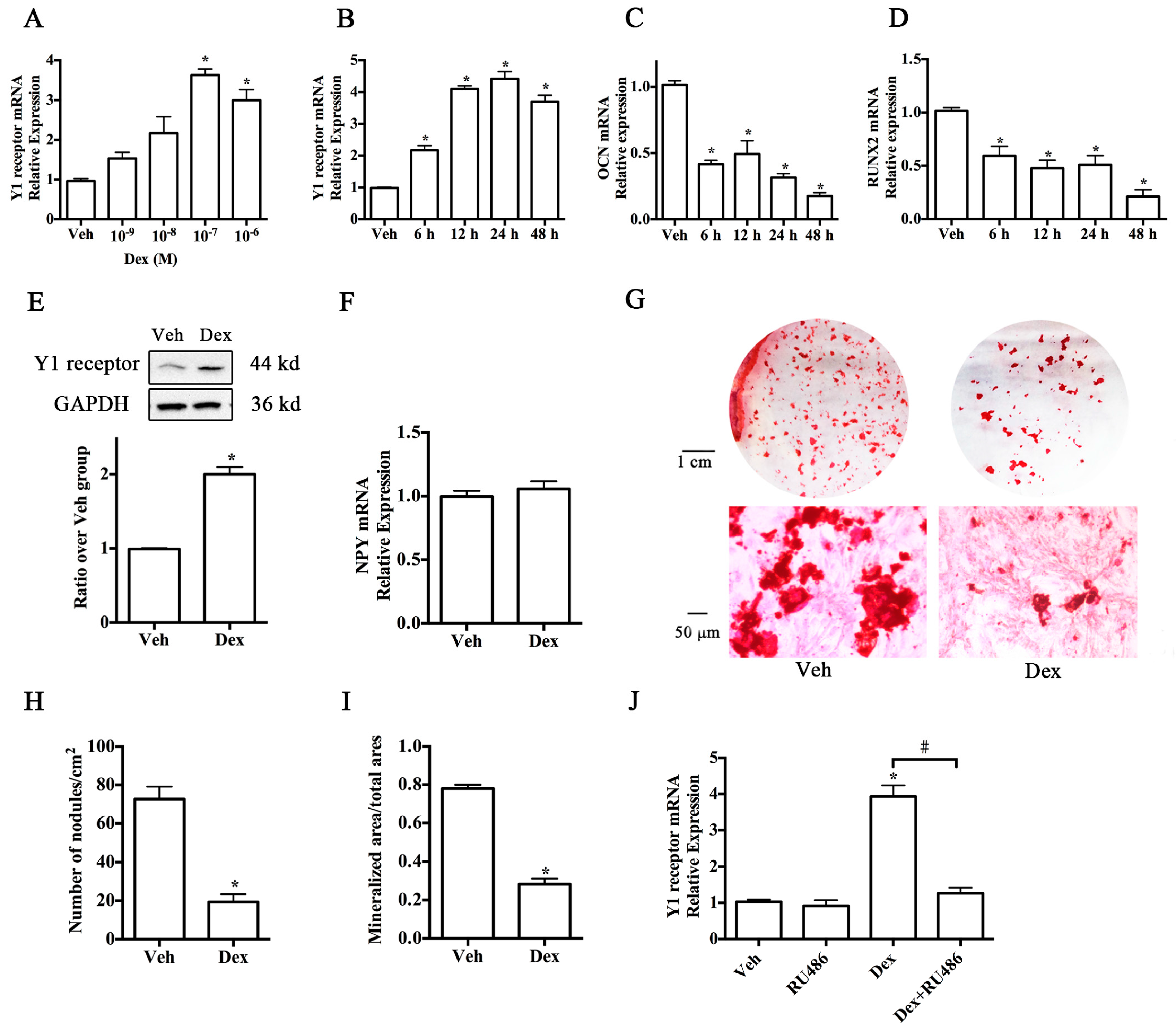
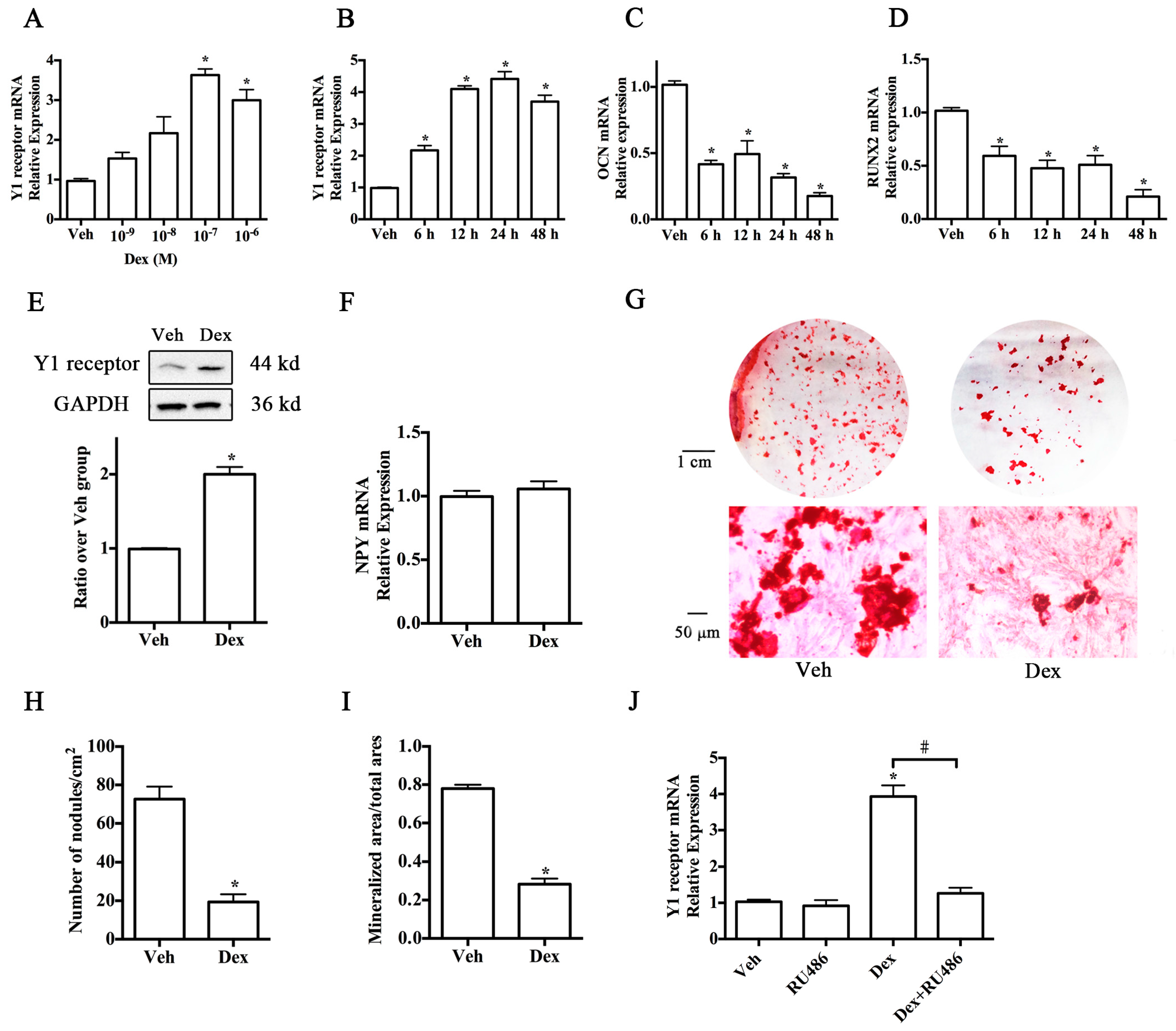
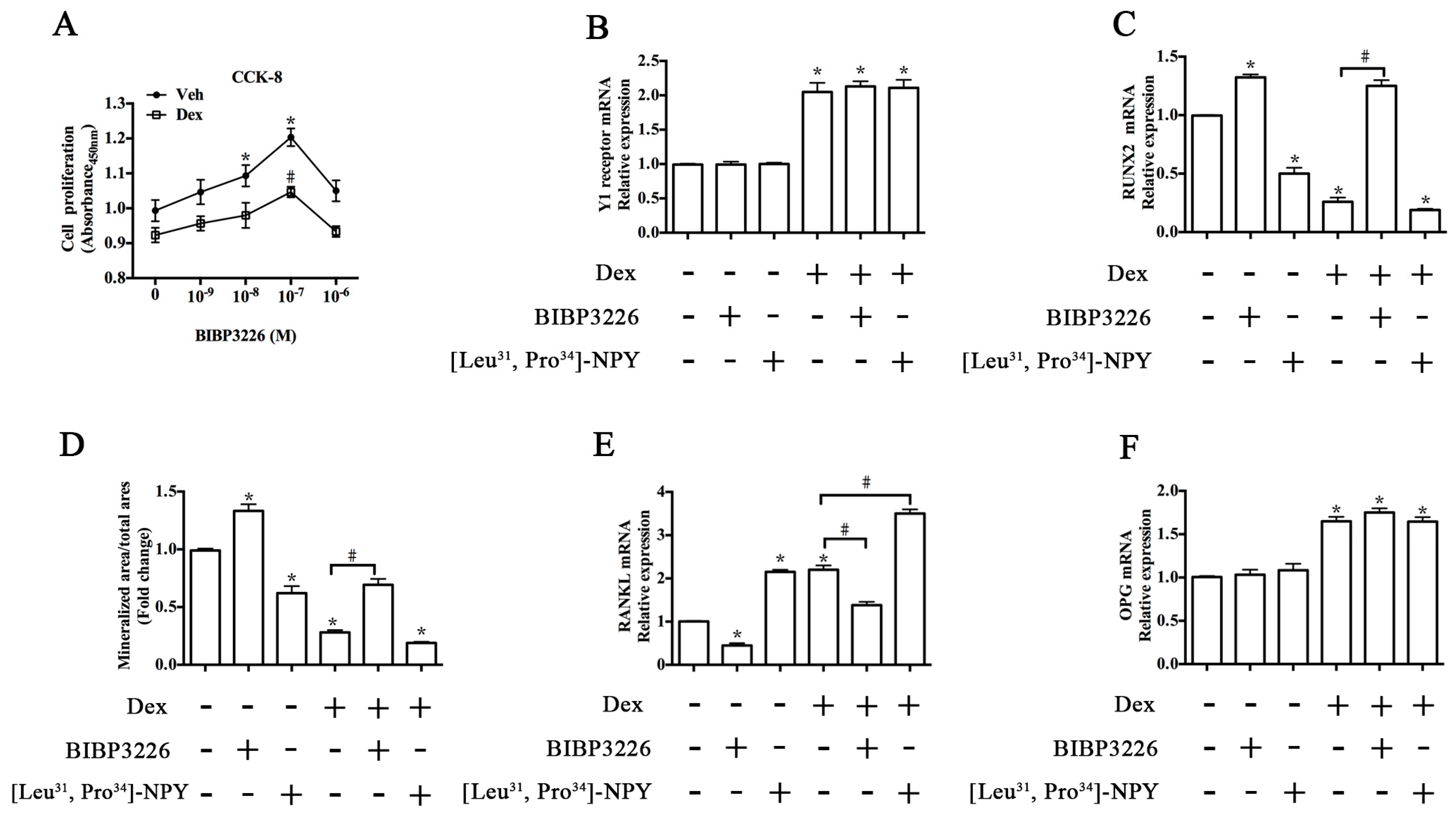
2.1. Upregulation of Y1 Receptor Expression by Dexamethasone
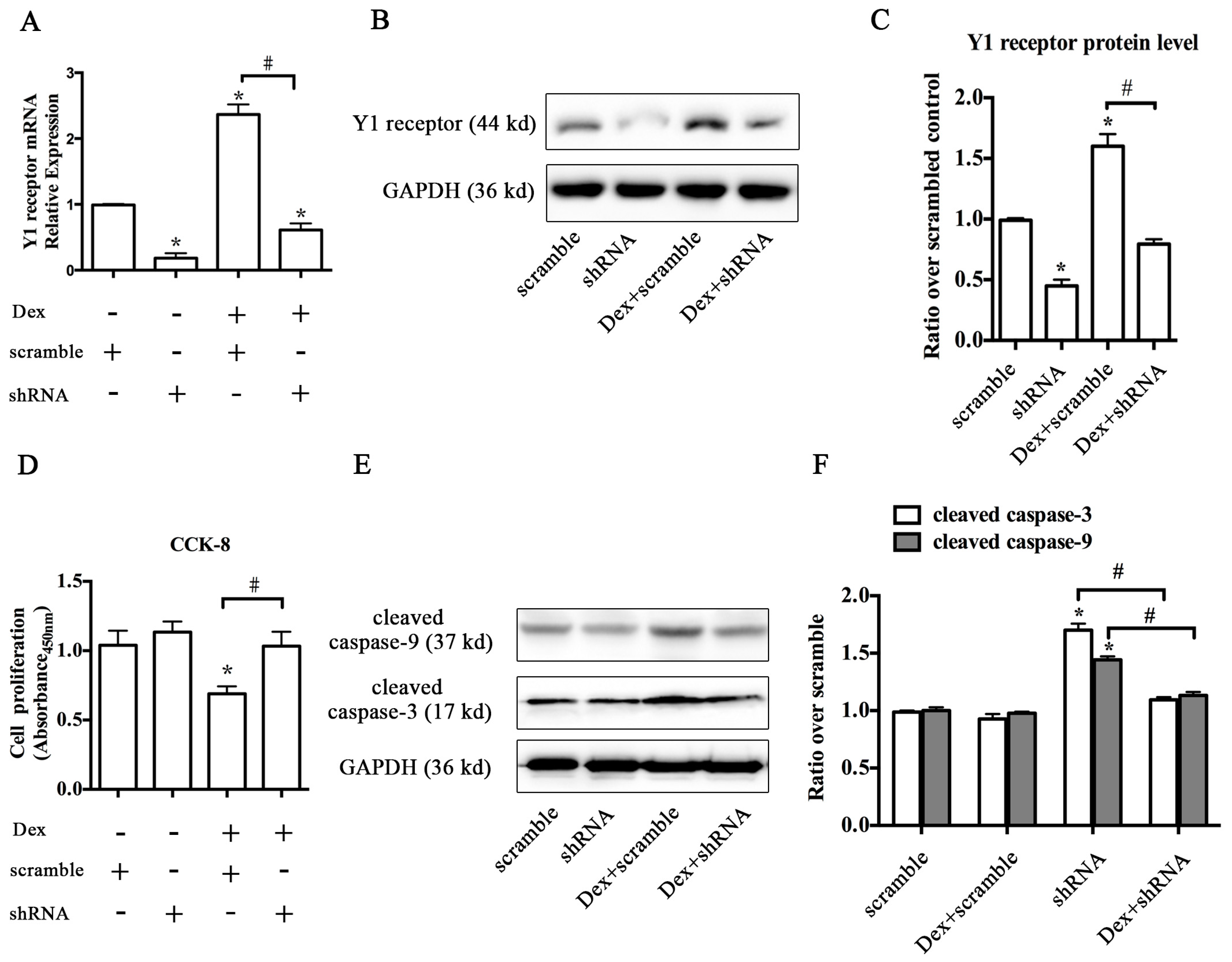
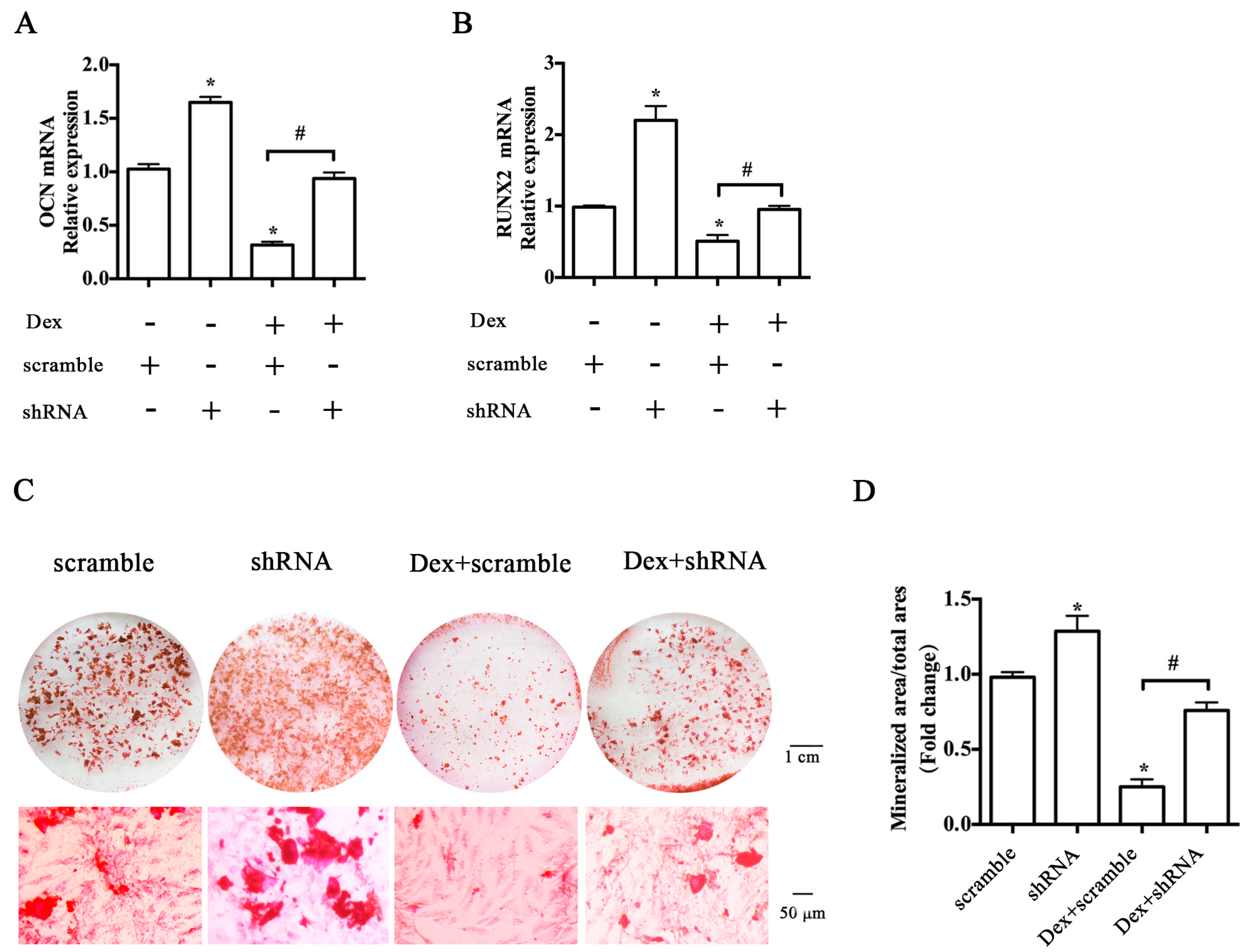
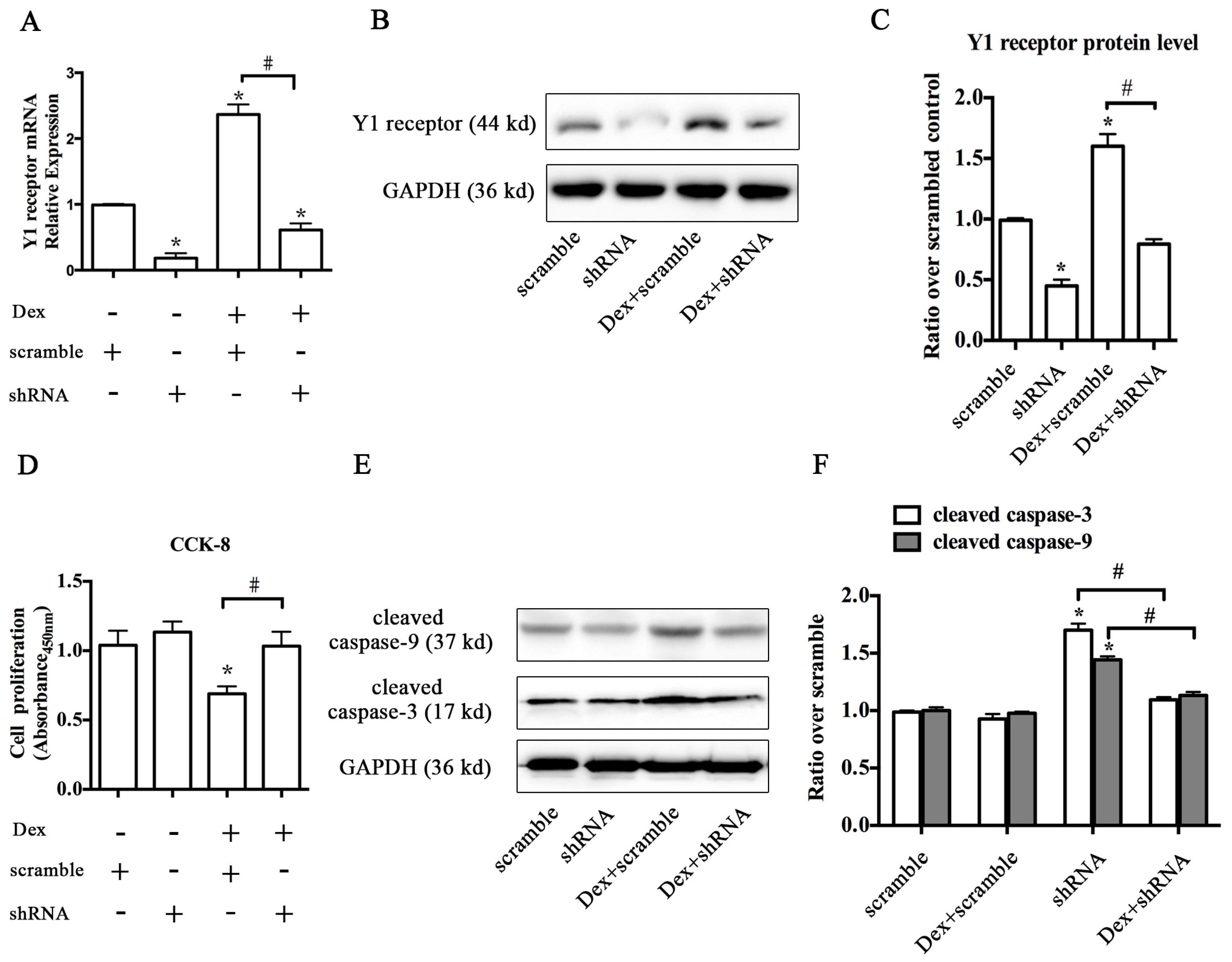
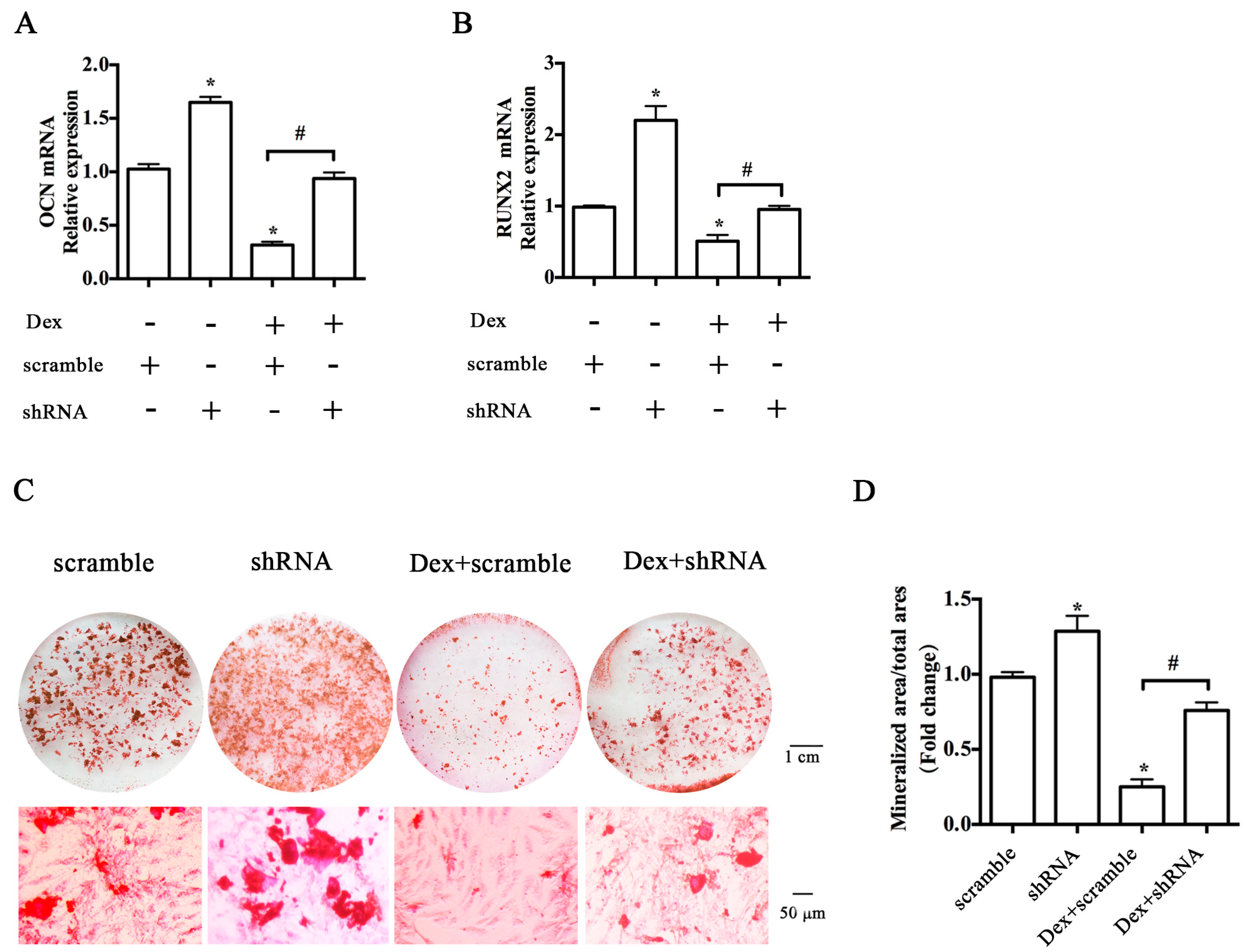
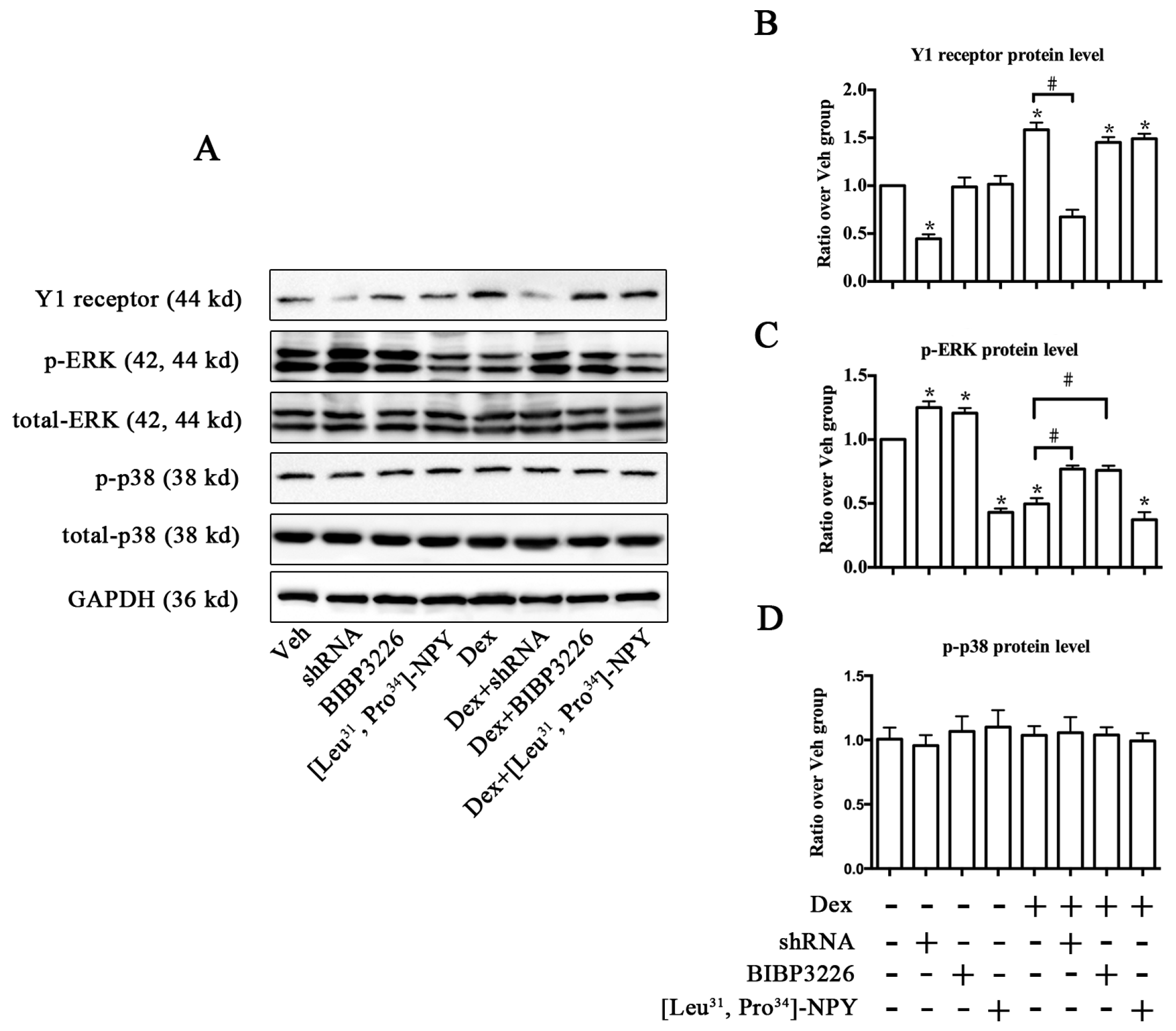
2.2. Knockdown of the Y1 Receptor Enhanced Osteoblast Differentiation
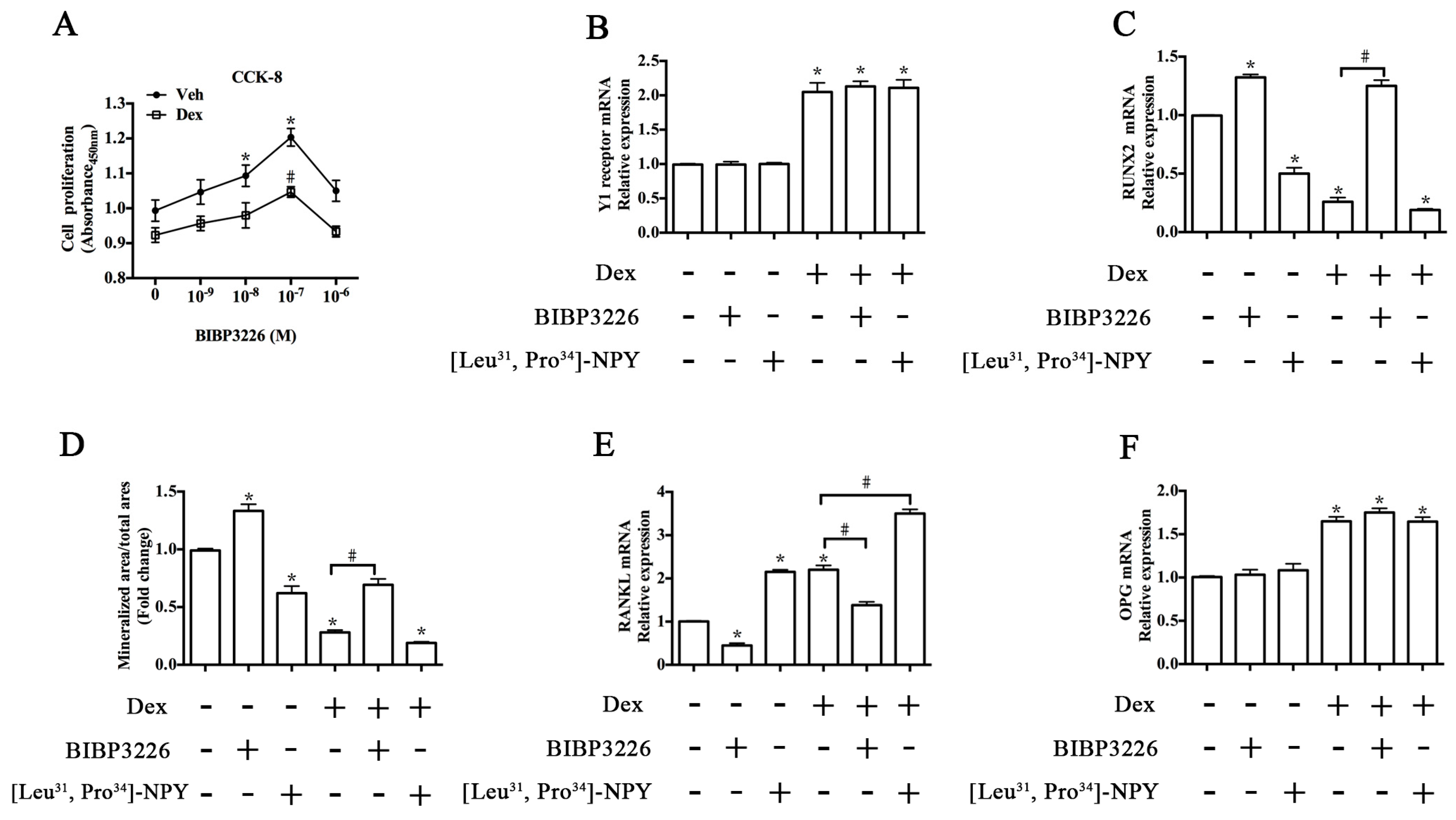
2.3. Y1 Receptor Antagonist Regulated the Mineralization of MC3T3-E1 Cells
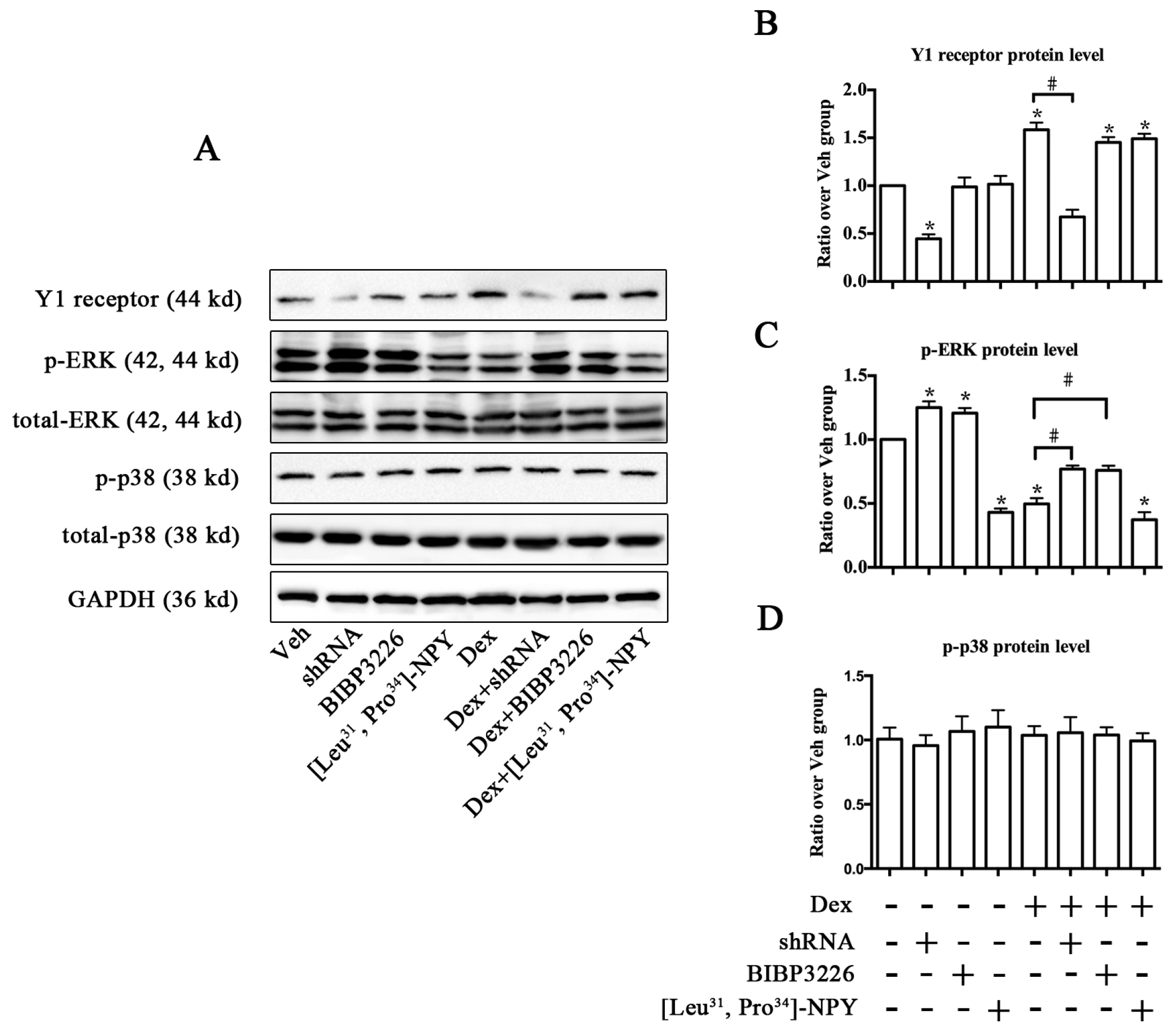
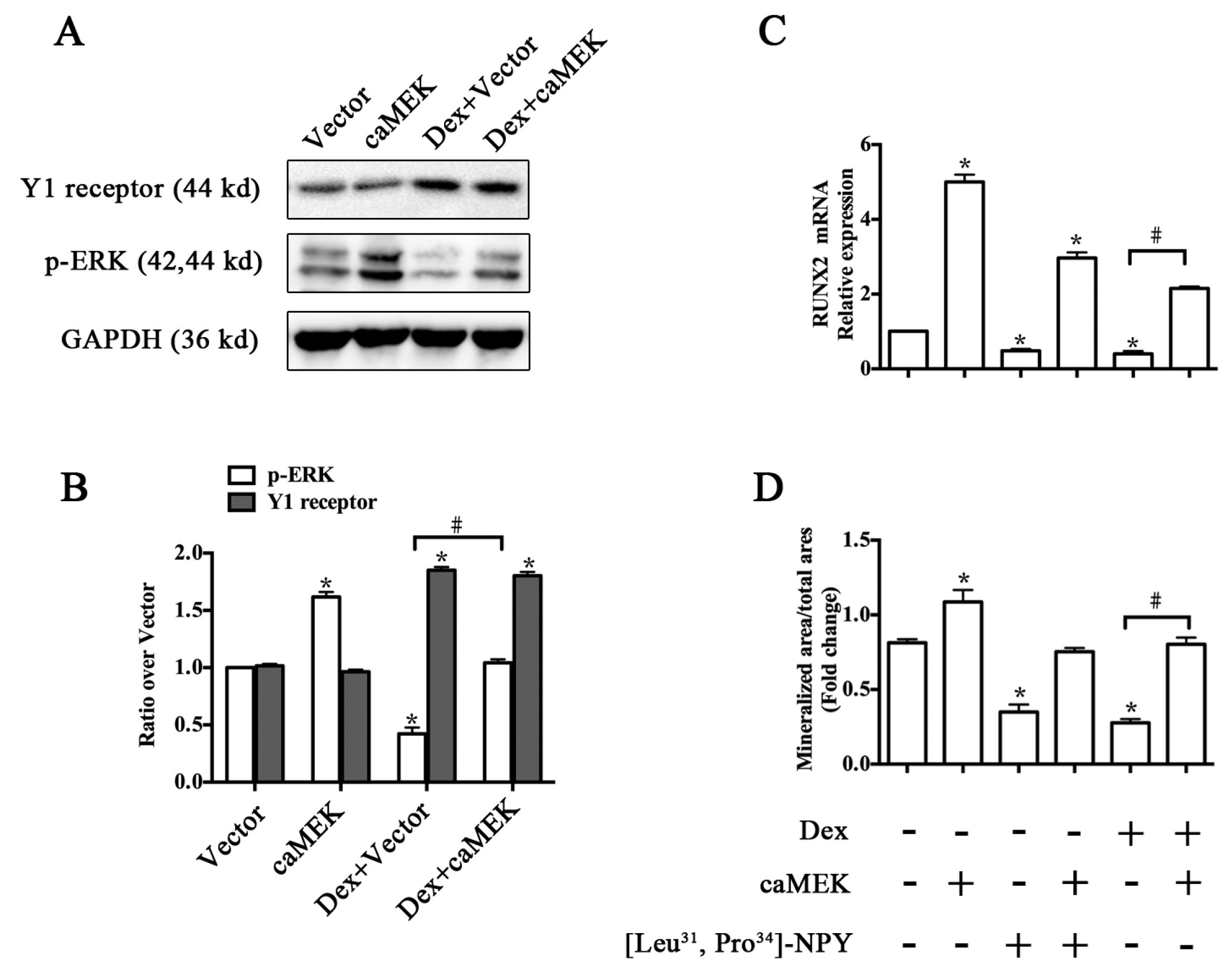
2.4. ERK Signaling Participated in Y1 Receptor-Mediated Suppression of Osteogenic Differentiation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Transfection of Y1 Receptor shRNA Plasmid
4.3. Transfection of Active Mutant of MEK-1 cDNAs
4.4. Cell Proliferation and Viability Assay
4.5. Real-Time PCR Analysis
4.6. Western Blot Analysis
4.7. Alizarin Red S Staining
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Compston, J. Management of glucocorticoid-induced osteoporosis. Nat. Rev. Rheumatol. 2010, 6, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Tait, A.S.; Butts, C.L.; Sternberg, E.M. The role of glucocorticoids and progestins in inflammatory, autoimmune, and infectious disease. J. Leukoc. Biol. 2008, 84, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, S.; Brama, M.; Fornari, R.; Greco, E.A.; Spera, G.; Malavolta, N. Glucocorticoid-induced osteoporosis: an osteoblastic disease. Aging Clin. Exp. Res. 2007, 19, 5–10. [Google Scholar] [PubMed]
- Weinstein, R.S.; Jilka, R.L.; Parfitt, A.M.; Manolagas, S.C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: Potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998, 102, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.; Sun, L.; Robinson, L.J.; Tourkova, I.L.; Liu, L.; Wang, Y.; Zhu, L.L.; Liu, X.; Li, J.; Peng, Y.; et al. ACTH protects against glucocorticoid-induced osteonecrosis of bone. Proc. Natl. Acad. Sci. USA 2010, 107, 8782–8887. [Google Scholar] [CrossRef] [PubMed]
- Henneicke, H.; Gasparini, S.J.; Brennan-Speranza, T.C.; Zhou, H.; Seibel, M.J. Glucocorticoids and bone: local effects and systemic implications. Trends Endocrinol. Metab. 2014, 25, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Kar, R.; Gluhak-Heinrich, J.; Yao, W.; Lane, N.E.; Bonewald, L.F.; Biswas, S.K.; Lo, W.K.; Jiang, J.X. Glucocorticoid-induced autophagy in osteocytes. J. Bone Miner. Res. 2010, 25, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, N.; Huang, X.; Xu, J.; Fernandes, J.C.; Dai, K.; Zhang, X. Dexamethasone shifts bone marrow stromal cells from osteoblasts to adipocytes by C/EBPalpha promoter methylation. Cell Death Dis. 2013, 4, e832. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.C.; Baldock, P.A. Central and peripheral mechanisms of the NPY system in the regulation of bone and adipose tissue. Bone 2012, 50, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Allison, S.J.; Lee, N.J.; Baldock, P.A.; Brouard, N.; Rost, S.; Enriquez, R.F.; Sainsbury, A.; Lamghari, M.; Simmons, P.; et al. Greater bone formation of Y2 knockout mice is associated with increased osteoprogenitor numbers and altered Y1 receptor expression. J. Biol. Chem. 2007, 282, 19082–19091. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.J.; Doyle, K.L.; Sainsbury, A.; Enriquez, R.F.; Hort, Y.J.; Riepler, S.J.; Baldock, P.A.; Herzog, H. Critical role for Y1 receptors in mesenchymal progenitor cell differentiation and osteoblast activity. J. Bone Miner. Res. 2010, 25, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Baldock, P.A.; Allison, S.J.; Lundberg, P.; Lee, N.J.; Slack, K.; Lin, E.J.; Enriquez, R.F.; McDonald, M.M.; Zhang, L.; During, M.J.; et al. Novel role of Y1 receptors in the coordinated regulation of bone and energy homeostasis. J. Biol. Chem. 2007, 282, 19092–19102. [Google Scholar] [CrossRef] [PubMed]
- Baldock, P.A.; Sainsbury, A.; Couzens, M.; Enriquez, R.F.; Thomas, G.P.; Gardiner, E.M.; Herzog, H. Hypothalamic Y2 receptors regulate bone formation. J. Clin. Investig. 2002, 109, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Baldock, P.A.; Lee, N.J.; Driessler, F.; Lin, S.; Allison, S.; Stehrer, B.; Lin, E.J.; Zhang, L.; Enriquez, R.F.; Wong, I.P.; et al. Neuropeptide Y knockout mice reveal a central role of NPY in the coordination of bone mass to body weight. PLoS ONE 2009, 4, e8415. [Google Scholar] [CrossRef] [PubMed]
- Sousa, D.M.; Baldock, P.A.; Enriquez, R.F.; Zhang, L.; Sainsbury, A.; Lamghari, M.; Herzog, H. Neuropeptide Y Y1 receptor antagonism increases bone mass in mice. Bone 2012, 51, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Igwe, J.C.; Jiang, X.; Paic, F.; Ma, L.; Adams, D.J.; Baldock, P.A.; Pilbeam, C.C.; Kalajzic, I. Neuropeptide Y is expressed by osteocytes and can inhibit osteoblastic activity. J. Cell. Biochem. 2009, 108, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.J.; Nguyen, A.D.; Enriquez, R.F.; Doyle, K.L.; Sainsbury, A.; Baldock, P.A.; Herzog, H. Osteoblast specific Y1 receptor deletion enhances bone mass. Bone 2011, 48, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Laborie, C.; Bernet, F.; Kerckaert, J.P.; Maubert, E.; Lesage, J.; Dupouy, J.P. Regulation of neuropeptide Y and its mRNA by glucocorticoids in the rat adrenal gland. Neuroendocrinology 1995, 62, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Myrsen-Axcrona, U.; Karlsson, S.; Sundler, F.; Ahren, B. Dexamethasone induces neuropeptide Y (NPY) expression and impairs insulin release in the insulin-producing cell line RINm5F: Release of NPY and insulin through different pathways. J. Biol. Chem. 1997, 272, 10790–10796. [Google Scholar] [PubMed]
- Wang, F.S.; Lian, W.S.; Weng, W.T.; Sun, Y.C.; Ke, H.J.; Chen, Y.S.; Ko, J.Y. Neuropeptide Y mediates glucocorticoid-induced osteoporosis and marrow adiposity in mice. Osteoporos. Int. 2016, 27, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Meng, Z. Expression of caspase and apoptotic signal pathway induced by sulfur dioxide. Environ. Mol. Mutagen. 2010, 51, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.M.; Chen, Y.C.; Hsu, M.H.; Tain, Y.L.; Huang, Y.H.; Tiao, M.M.; Li, S.W.; Huang, L.T. Melatonin Alleviates Liver Apoptosis in Bile Duct Ligation Young Rats. Int. J. Mol. Sci. 2016, 17, 1365. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Koh, A.J.; Datta, N.S.; Zhang, J.; Keller, E.T.; Xiao, G.; Franceschi, R.T.; D’Silva, N.J.; McCauley, L.K. Impact of the mitogen-activated protein kinase pathway on parathyroid hormone-related protein actions in osteoblasts. J. Biol. Chem. 2004, 279, 29121–29129. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, E.N.; Ferrari, S.L. Beta-arrestin2 regulates parathyroid hormone effects on a p38 MAPK and NFkappaB gene expression network in osteoblasts. Bone 2009, 45, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Horsnell, H.; Baldock, P.A. Osteoblastic Actions of the Neuropeptide Y System to Regulate Bone and Energy Homeostasis. Curr. Osteoporos. Rep. 2016, 14, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Seitz, S.; Baschant, U.; Schilling, A.F.; Illing, A.; Stride, B.; Kirilov, M.; Mandic, V.; Takacz, A.; Schmidt-Ullrich, R.; et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010, 11, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, L.C.; Zeitz, U.; Schoppet, M.; Skalicky, M.; Schuler, C.; Stolina, M.; Kostenuik, P.J.; Erben, R.G. Prevention of glucocorticoid-induced bone loss in mice by inhibition of RANKL. Arthritis Rheum. 2009, 60, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Miguel, S.M.; Namdar-Attar, M.; Noh, T.; Frenkel, B.; Bab, I. ERK1/2-activated de novo Mapkapk2 synthesis is essential for osteogenic growth peptide mitogenic signaling in osteoblastic cells. J. Biol. Chem. 2005, 280, 37495–37502. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.W.; Lin, T.P.; Ko, J.Y.; Yeh, D.W.; Chen, M.W.; Ke, H.C.; Wu, S.L.; Wang, F.S. Cannabinoid receptor 1 regulates ERK and GSK-3beta-dependent glucocorticoid inhibition of osteoblast differentiation in murine MC3T3-E1 cells. Bone 2011, 49, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Pellieux, C.; Sauthier, T.; Domenighetti, A.; Marsh, D.J.; Palmiter, R.D.; Brunner, H.R.; Pedrazzini, T. Neuropeptide Y (NPY) potentiates phenylephrine-induced mitogen-activated protein kinase activation in primary cardiomyocytes via NPY Y5 receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.R.; Kim, C.W. Neuropeptide Y promotes beta-cell replication via extracellular signal-regulated kinase activation. Biochem. Biophys. Res. Commun. 2004, 314, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Lecat, S.; Belemnaba, L.; Galzi, J.L.; Bucher, B. Neuropeptide Y receptor mediates activation of ERK1/2 via transactivation of the IGF receptor. Cell Signal. 2015, 27, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, M.D.; Wolf, I.M.; Sanchez, E.R.; Witchel, S.F.; DeFranco, D.B. Glucocorticoid receptor physiology. Rev. Endocr. Metab. Disord. 2007, 8, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zheng, X.F.; Yang, Y.H.; Li, B.; Wang, Y.R.; Jiang, S.D.; Jiang, L.S. LGR4 acts as a key receptor for R-spondin 2 to promote osteogenesis through Wnt signaling pathway. Cell Signal. 2016, 28, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, E.; Boucher, M.J.; Mongrain, S.; Boudreau, F.; Asselin, C.; Rivard, N. Constitutive activation of the MEK/ERK pathway inhibits intestinal epithelial cell differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Gopalbhai, K.; Jansen, G.; Beauregard, G.; Whiteway, M.; Dumas, F.; Wu, C.; Meloche, S. Negative regulation of MAPKK by phosphorylation of a conserved serine residue equivalent to Ser212 of MEK1. J. Biol. Chem. 2003, 278, 8118–8125. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Primer Sequence (5′-3′) Sense/Antisense | GenBank Number |
|---|---|---|
| Y1 Receptor [10] | CTCGCTGGTTCTCATCGCTGTGGAACGG | NM_010934 |
| GCGAATGTATATCTTGAAGTAG | ||
| NPY | CTCGTGTGTTTGGGCATTC | NM_023456 |
| TAGTGTCGCAGAGCGGAGTA | ||
| RUNX2 [34] | GACGAGGCAAGAGTTTCACC | NM_009820 |
| GGACCGTCCACTGTCACTTT | ||
| OCN | CAAGCAGGGAGGCAATAAGG | NM_007541 |
| CGTCACAAGCAGGGTTAAGC | ||
| OPG [34] | AGCTGCTGAAGCTGTGGAA | NM_008764 |
| GGTTCGAGTGGCCGAGAT | ||
| RANKL [34] | GAAGGCTCATGGTTGGATGT | NM_011613 |
| GTAGCCCAAGGGTATTTCAG | ||
| β-actin [34] | TGACAGGATGCAGAAGGAGA | NM_007393 |
| CGCTCAGGAGGAGCAATG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.; Zhu, C.; Xu, W.; Jiang, L.; Jiang, S. Neuropeptide Y1 Receptor Regulates Glucocorticoid-Induced Inhibition of Osteoblast Differentiation in Murine MC3T3-E1 Cells via ERK Signaling. Int. J. Mol. Sci. 2016, 17, 2150. https://doi.org/10.3390/ijms17122150
Yu W, Zhu C, Xu W, Jiang L, Jiang S. Neuropeptide Y1 Receptor Regulates Glucocorticoid-Induced Inhibition of Osteoblast Differentiation in Murine MC3T3-E1 Cells via ERK Signaling. International Journal of Molecular Sciences. 2016; 17(12):2150. https://doi.org/10.3390/ijms17122150
Chicago/Turabian StyleYu, Wei, Chao Zhu, Wenning Xu, Leisheng Jiang, and Shengdan Jiang. 2016. "Neuropeptide Y1 Receptor Regulates Glucocorticoid-Induced Inhibition of Osteoblast Differentiation in Murine MC3T3-E1 Cells via ERK Signaling" International Journal of Molecular Sciences 17, no. 12: 2150. https://doi.org/10.3390/ijms17122150