1. Introduction
In 1951, Pauling and colleagues first defined two main secondary elements (α-helix and β-sheet) based on the intra-backbone hydrogen bond patterns in proteins [
1]. They correctly detected the idealized π-helix but incorrectly predicted that 3
10-helix would not occur due to unfavorable angles. However, approximately 4% of residues in proteins have been shown to occur in this secondary element [
2]. Except for the two predominant secondary structure elements and two helical elements, other minor secondary structural elements (SSE) such as β-turns [
3], β-bulges [
4], γ-turns [
5] and loops have been defined using the hydrogen bond information in proteins. All SSEs are usually grouped into three larger classes: helix, strand and coil [
6]. To date, secondary structures have been extensively employed in structure visualization [
7], classification [
8], comparison [
9], and prediction [
10].
The first SSE assignment program, proposed by Levitt and colleagues, automatically detected SSEs using C
α distance, inter-C
α torsion angle and peptide hydrogen bond patterns [
11]. DSSP was subsequently developed and has become the most popular program in the field, serving as the “gold standard” [
12]. Moreover, most SSE prediction methods are based on DSSP assignments [
13], which identifies backbone hydrogen bond patterns based on an electrostatic approximation of hydrogen bond energy followed by SSE assignment using hydrogen bond pattern information. STRIDE, which is the second most popular algorithm at present, employs a modified hydrogen bond energy function and the statistical probability factors of main-chain dihedral angles derived from Protein Data Bank (PDB) [
14] records to perform SSE assignments [
15]. SECSTR is a new addition to the DSSP program that is dedicated to identifying π-helices, which were seldom assigned by older versions of DSSP and STRIDE [
16].
In addition to the aforementioned programs, which assign SSEs by detecting hydrogen bond information between backbone atoms, more than a dozen geometry-based SSE assignment programs have been developed. Geometry-based secondary structure assignment programs can be generally categorized into two groups: (1) methods that use the geometrical restraint of local fragments and (2) methods that fit C
α coordinates to a line or curve. P-SEA uses a short-range C
α distance mask (
i to
i + 2,
i + 3 and
i + 4) and two dihedral angle criteria for secondary structure assignment [
6]. KAKSI develops an assignment by defining allowed C
α distance measures and dihedral angles [
17]. Similar to P-SEA, XTLSSTR also calculates three distances and two backbone dihedral angles to determine SSE, but two distances are H-bond distances instead of C
α distances [
18]. PALSSE delineates SSEs from C
α coordinates and uses distance as well as torsion angle restraints to detect core elements; core elements are then extended to longer fragments [
19]. SABA introduces a novel geometrical parameter, a pseudo center, which is the midpoint of two continuous C
αs, and assigns SSEs using cut-off criteria for distances as well as dihedral angles of two or more pseudo centers and C
α atoms [
20]. PROSS defines SSEs based solely on backbone torsion angles [
21], whereas SENGO uses the angle between successive peptide bonds for helix assignment and backbone dihedrals as well as alternating peptide bonds for β-sheet assignment [
22]. More recently, DISICL and PCASSO have been developed. DISICL classifies SSEs into 18 distinct classes based solely on the main-chain dihedral angles of two consecutive residues; PCASSO applies Random Forests in learning 258 geometric features calculated by C
αs and pseudo centers (see SABA) at different positions [
23,
24].
Several other programs can be classified into the second category. DEFINE assigns SSEs by matching C
α coordinates with a linear distance matrix of ideal secondary structures [
25]. STICK, which is considered a variant of DEFINE, fits a set of line segments independent of any external secondary structure definition to avoid the problem of fitting a single line to a bent structure [
26]. SSE assignment in P-CURVE is based on matching a peptide backbone to motifs that have idealized helical parameters and generates a global curved axis [
27]. In particular, SKSP and PSSC do not belong to any category mentioned before: SKSP performs SSE assignments by averaging four popular programs: STRIDE, KAKSI, SECSTR and PSEA [
28]; PSSC uses DSSP output and introduces detailed eight-character secondary structure information to characterize protein structures [
29].
In general, the majority of geometry-based methods exhibit a broad consensus at most helix and strand core segments in proteins. For KAKSI, the agreement with DSSP is 91.7% and 92.1% for helices and strands, respectively, whereas the agreement between P-SEA and DSSP for the two major elements is 93.8% and 78.4% [
6,
17]. The main difficulties for secondary structure assignment can be categorized into three areas: (1) locating the terminus of the helix/strand; (2) distinguishing distortions and breaks in the secondary structure [
17]; and (3) detecting and prioritizing subtle secondary structures, such as 3
10-helices and π-helices. As DSSP recognizes SSEs well and agrees with intuitive visual criteria [
15], irregular and outlier fragments assigned by DSSP need to be distinguished, and the remaining “regular” fragments may serve as templates for new SSE assignments to make the assignments more uniform and visually acceptable. To address this problem, we developed a method SACF that assigns SSEs in three steps: First, outlier SSE fragments are detected. Next, the central fragments are derived by clustering the remaining fragments. Finally, new SSE fragments are assigned by aligning them to the template central fragments. An outlier SSE fragment is one that is far away from its
k-nearest neighbor fragments. SSE fragments are often closely packed together. Thus, an outlier SSE fragment is irregular compared with its neighbors. A central SSE fragment is a fragment that has the minimum total RMSD compared to all other fragments within a cluster. Instead of only excluding local outlier torsional angles (ϕ/ψ) as STRIDE does [
15], our method focuses on whole C
α fragments and addresses irregular SSEs. Several methods have been proposed for capturing outliers [
30] and performing data clustering [
31]. In the present study, a geometric clustering algorithm [
32] proposed by us was applied to the clustering process, whereas a local distance-based outlier factor (
LDOF) was used in the outlier fragment detection process [
33]. The central fragment in each cluster served as a template fragment, and accurate assignment to a particular type is made based on a smaller root-mean square deviation (RMSD) than the threshold after alignment to the template fragment. We assumed that the best method should uniformly assign secondary structures, meaning that the same secondary structures should be aligned with minimum RMSD. Our method does not utilize hydrogen bonds, backbone dihedral angles, backbone NH or CO coordinates, or virtual bond lengths or angles. The program SACF is available upon request.
More than 20 SSE assignment methods have been developed; however, only Martin
et al. undertook a comparison for six SSE assignment methods [
17] and Colloc’h compared three methods: DSSP, P-CURVE and DEFINE [
34]. Moreover, the agreement measures were inconsistent across different papers. We applied our algorithm to identify helices and β-sheets in the protein set and compared our assignments with 10 available programs that employ different criteria for SSE assignment: DSSP, STRIDE, P-SEA, KAKSI, DISICL, PALSSE, SEGNO, PROSS, XTLSSTR and PCASSO. The comparisons were performed based on two X-ray protein databases with middle and low resolution, as well as with NMR protein structures. We also discuss the N and C cap region of different SSE assignment methods, as most disagreements between different methods arise in the terminal regions of the assigned SSEs [
13,
28,
34].
2. Results and Discussion
Set
T consists of 2817 structures with resolutions between 2.0 and 3.0 Å, which was selected to compare our method with ten other programs, including two hydrogen bond-based SSE assignment programs (DSSP and STRIDE) and nine geometry-based methods. As shown in
Table 1, twelve pairs of programs share a
Q3 score of more than 84% (bold). The agreement between the nine geometry-based methods and two hydrogen bond-based methods ranged from 72.9% to 93.5%, whereas the range of agreement among the geometry-based methods was wider, from 63.1% to 86.2%. Notably, all of the SSEs are generally grouped into three categories (helix, strand, and coil) because most geometry-based methods do not provide subtle secondary structure types. In summary, SACF agrees better with DSSP and STRIDE (84.7% and 85.1% respectively) than with other geometry-based methods except PCASSO. PCASSO achieves high agreement with DSSP (93.5%) because the protein secondary structures in the training set were assigned by DSSP and 258 geometric features were used in random decision forests. KAKIS and PROSS have similar
Q3 scores with DSSP; the agreement between these two methods and DSSP is 83.5% and 84.3%. DISICL and PALSSE assignment results are very different from the other methods. We also provide a comparison of the 11 methods on set
L and set
N (
Tables S1 and S2); the results show that these methods share similar
Q3 scores with DSSP on set
L, except for PCASSO, with a
Q3 score of 93.5% on set
T and a
Q3 score of 88.1% on set
L. Konagurthu reported that the agreement of β-strand between DSSP and STRIDE for NMR proteins was rather poor [
13]; however, we found that these two methods show similar agreement with β-strands for the NMR structures.
SOV scores are usually employed to evaluate secondary structure predictions, but this criterion can also be applied between two structure assignments [
17]. The
SOV score value is dependent on which method is selected as the reference assignment result; we take each method as the reference in turn. As shown in
Table 2 and
Table 3, we computed
SOV scores between any two of the 11 SSE assignment methods for helix and β-sheet.
For helix comparison, when the SACF assignment result is taken as the reference, the highest SOV score is obtained with DSSP (96.6%), followed by PCASSO (95.2%). If the DSSP assignment result is taken as the reference, PCASSO achieves an SOV score of 94.1% compared with DSSP, with an SOV score of 93.7% between STRIDE and DSSP. SACF yields an SOV score of 91.3% with DSSP, while KAKSI and PROSS show similar SOV scores with DSSP compared with SACF. When DISICL and PALSSE are selected as references, the SOV scores between other methods and these two methods are relatively low, ranging from 72.8% to 89.9% for DISICL and from 47.8% to 69.0% for PALSSE.
For β-sheet segment comparison,
SOV scores are lower compared with helix, as β-sheets are more irregular than helices [
34]. SACF, KAKSI, SEGNO, and PCASSO achieve
SOV scores of 81.2%, 88.0%, 80.4% and 89.2%, respectively, compared with DSSP as the reference method. For a given reference assignment in SACF, the
SOV scores between SACF and four methods (DSSP, STRIDE, KAKSI, PCASSO) are very close. Similar to helix, DISICL and PALSSE show very poor
SOV scores compared with the other methods.
In conclusion, SACF, KAKSI, and PROSS show similar agreement with DSSP, while a higher agreement is seen between PCASSO and DSSP. Among the four methods SACF, KAKSI, PROSS and PCASSO, only SACF divides helix into three sub secondary elements: α-helix, 310-helix, π-helix and left-handed helix. The aim of SACF is to make the secondary structure elements more uniform, and every element has its unique Cα fragment conformation; thus, some irregular β-sheet elements assigned by DSSP, such as β-bulge and β-hairpin, are selected as outliers by the outlier detection process of our algorithm, as these elements are short, rare and have similar Cα conformations with other elements such as loops and turns in proteins.
The length distributions of helices and strands assigned by SACF, DSSP, STRIDE, P-SEA, KAKSI, DISICL, and PALSSE on set
T are shown in
Figure 1. The average number of residues are 10.19 (SACF), 9.31 (DSSP), 9.61 (STRIDE), 11.64 (P-SEA), 12.53 (KAKSI), 5.90 (DISICL), and 13.67 (PALSSE) for helix and 4.69 (SACF), 5.38 (DSSP), 5.36 (STRIDE), 6.38 (P-SEA), 5.88 (KAKSI), 3.05 (DISICL), and 9.32 (PALSSE) for strand in β-sheet. DISICL assigns a large number of 1-residue-long helices (11,605) and 1-residue-long strands in β-sheet (16,123), which are not shown in
Figure 1. The distribution of the number of residues per helix has a jagged curve around 4 or 5 residues, except for DISICL and KAKSI. KAKSI provides the second highest number of long helices (more than 15 residues), while SACF, DSSP, STRIDE, and P-SEA assign very similar length distributions for helices of more than 12 residues. SACF assignment results in a slightly smaller number of 3-residue-long helices than both DSSP and STRIDE, whereas P-SEA and KAKSI do not assign helices shorter than 5 residues.
In the β-strand distribution, SACF assigns a larger number of strands with 2 to 3 residues than DSSP and STRIDE, as we provide a β-sheet ladder matching step for single strands. In the range of 4 to 7 residues, small differences are observed between SACF, DSSP and STRIDE; however, P-SEA and KAKSI show larger numbers of SSEs in this scope. For the zone of more than 8 residues in length, PALSSE assignment results in the largest number of strands in β-sheet, followed by P-SEA. In this range (length >8 residues), DSSP and STRIDE assign more strands in β-sheet than does SACF.
The capping regions show the most differences between different SSE assignment methods [
17]. If we take the cap regions defined by DSSP as the standard, we search the positions corresponding to the N and C caps of DSSP with other methods. Analyses of the N and C caps defined by DSSP and other methods are shown in
Table 4 and
Table 5. Seven methods, including STRIDE, SACF, P-SEA, KAKSI, SEGNO, PROSS, and PCASSO, have an overall agreement of more than 80% with DSSP, but the number of helices identical to DSSP are diverse. STRIDE assignment results in 11,388 helices identical to DSSP, as they both apply a hydrogen bond pattern in SSE assignment. P-SEA and KAKSI only have 1639 and 1761 helices, respectively, that are identical to the DSSP assignment results, while these numbers for SACF and PCASSO are 5194 and 5950, respectively. P-SEA, KAKIS and SEGNO tend to extend the C cap and N cap compared with DSSP assignment. By contrast, SACF and PCASSO prefer to reduce both cap regions.
Compared with assigning the extremities of helices, the N cap and C cap of β-sheet assigned by other methods (except STRIDE) are more inconsistent with DSSP. Similar to helix, SACF and PCASSO prefer to reduce both the N and C cap regions by one or two residues compared with DSSP, whereas P-SEA, KAKSI and SEGNO are more likely to add one or two residues to both terminals of helices and β-sheets defined by DSSP. The residues located in the cap region defined by DSSP but reduced by SACF indicate that the Cα fragments of these residues are irregular and detected as outliers although their backbone atoms can form hydrogen bonds in the DSSP SSE assignment standard.
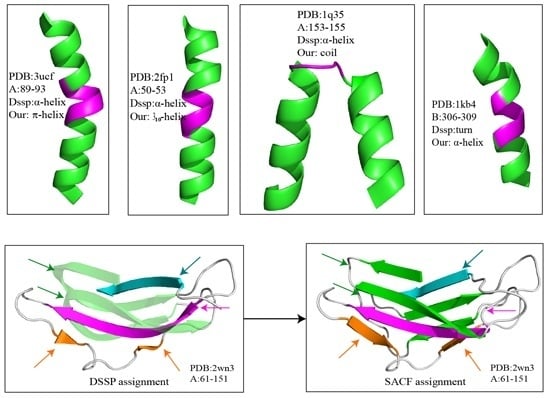
Figure 2 shows several examples of disagreement between our method and DSSP. The agreement between our method and DSSP for π-helices is better than that for 3
10-helices; the π-helices we assigned were more uniform, and their geometry differed from that of α-helices (
Figure 2a and
Figure 3). The top four panels of
Figure 2 illustrate the subtle differences in helix assignment. Although 3
10-helices are not easily distinguished from α-helices because their C
α-fragment poses are so similar, we continued to be able to identify fragments that should only match 3
10-helices (
Figure 2b). Specifically, the 3
10-helix-forming (
i,
i + 3) hydrogen bond energy is also stronger than the α-helix-forming (
i,
i + 4) hydrogen bond energy at this fragment according to the DSSP output (Figure S1). The C
α fragments of three helices (α-helix, 310-helix and π-helix) assigned by SACF are more uniform and can be clearly separated, whereas the C
α fragments of the three helices assigned by DSSP show some intersection (
Figure 3).
Figure 2c,d describe the disagreement in α-helix assignment. Because the merging process and kink pose in our method are selected based on their incidence in the DSSP assignment, a long helix assigned by DSSP is divided into two individual helices in our assignment (
Figure 2c), and two helices assigned by DSSP are “merged” into a single helix because the fragment between the two helices can be matched to our central helix poses.
The bottom two panels in
Figure 2 show examples of the disagreement in β-sheet assignment between our method and DSSP. Our method often splits kinked β-strands or β-strands accompanied by β-bulges assigned by DSSP into two or more structures because the curved part of the β-strand does not match our central β-strand poses. The residues establish hydrogen bonds with their pairs but do not match the β-strand central poses.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}