
Chemical Conditioning as an Approach to Ischemic Stroke Tolerance: Mitochondria as the Target


Abstract
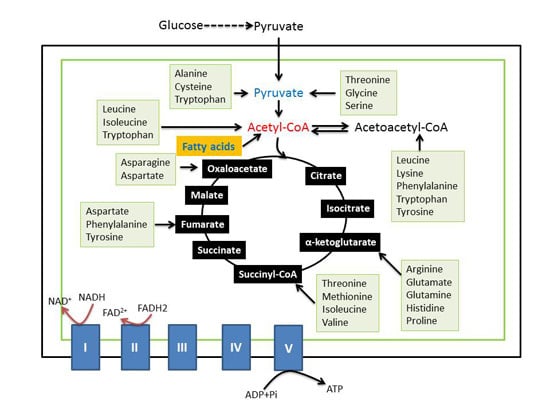
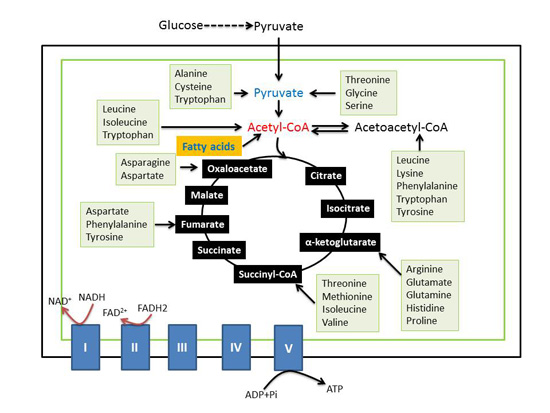
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
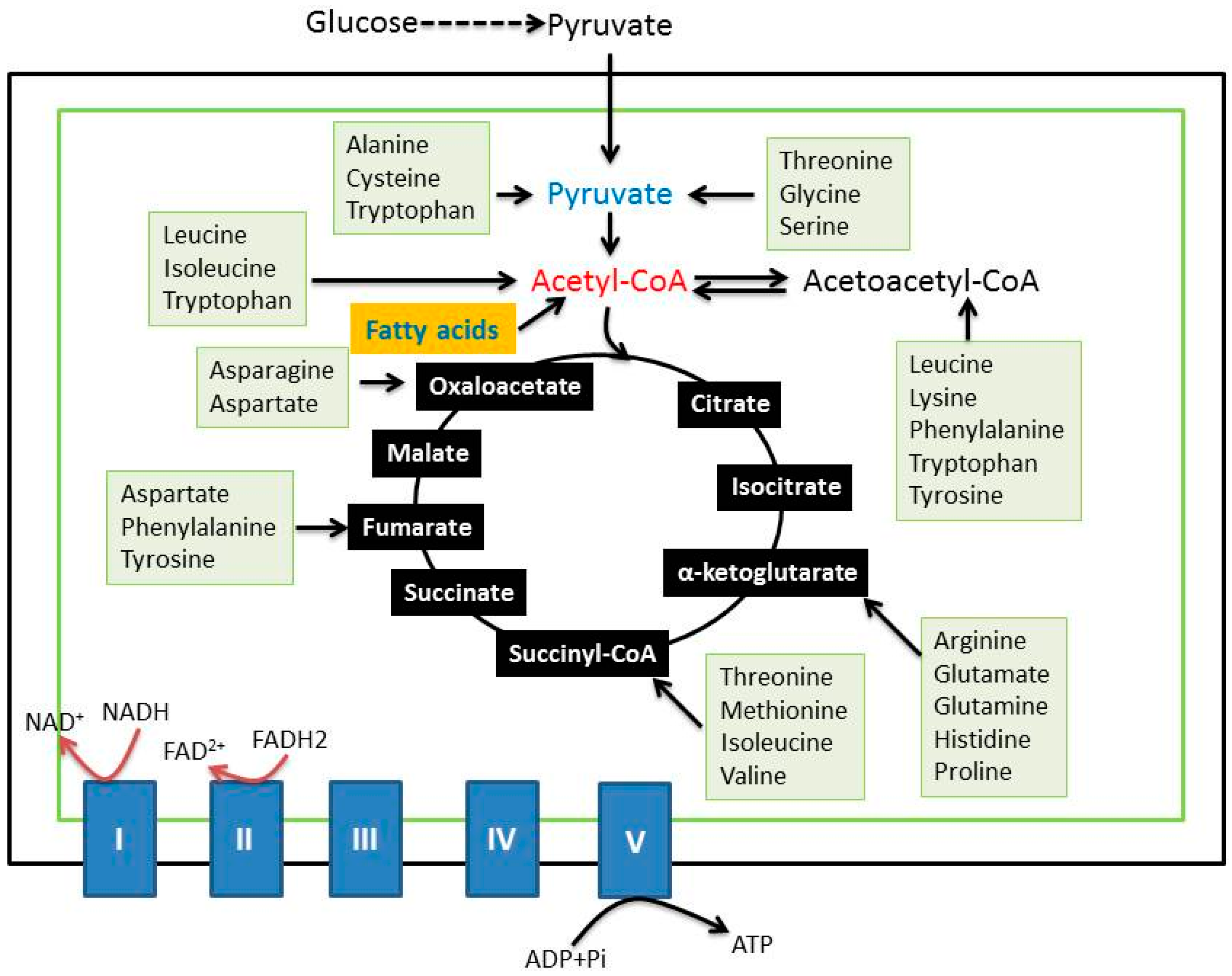{kind=link}
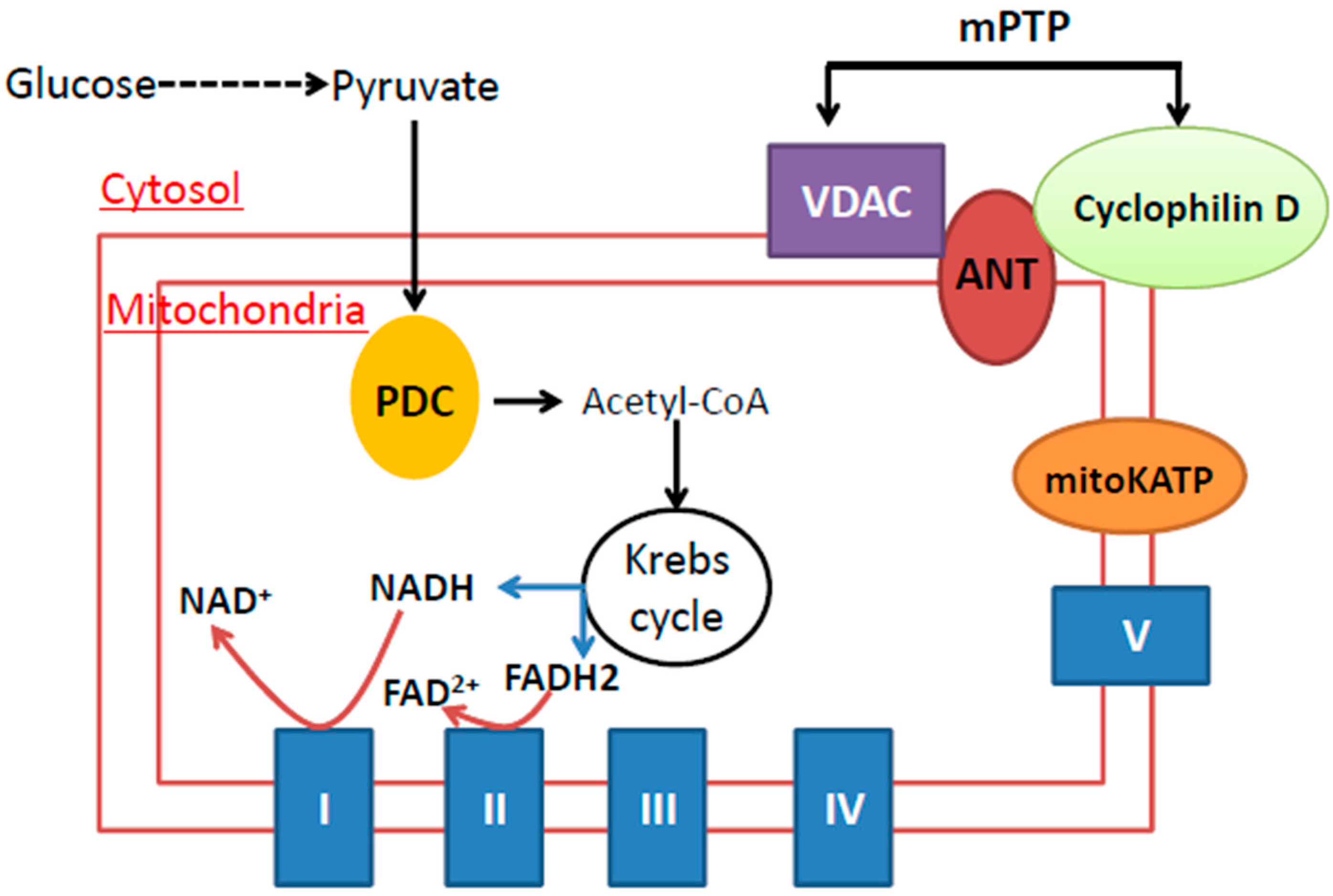{kind=link}
1. Introduction
2. Mitochondria and Ischemic-Reperfusion Injury
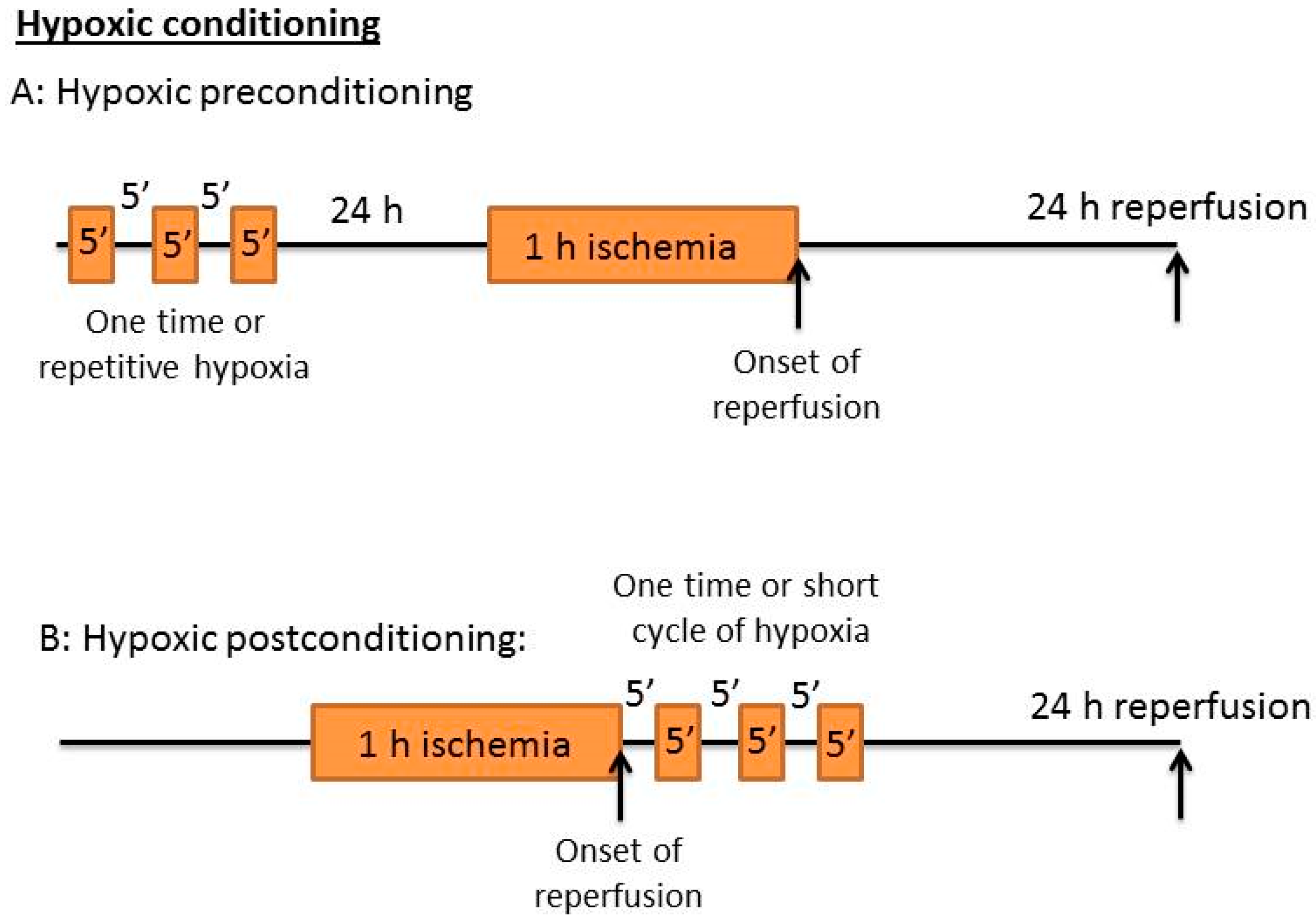
3. Ischemic Conditioning: Ischemic Pre-C and Post-C
4. Hypoxic Conditioning: Hypoxic Pre-C and Post-C
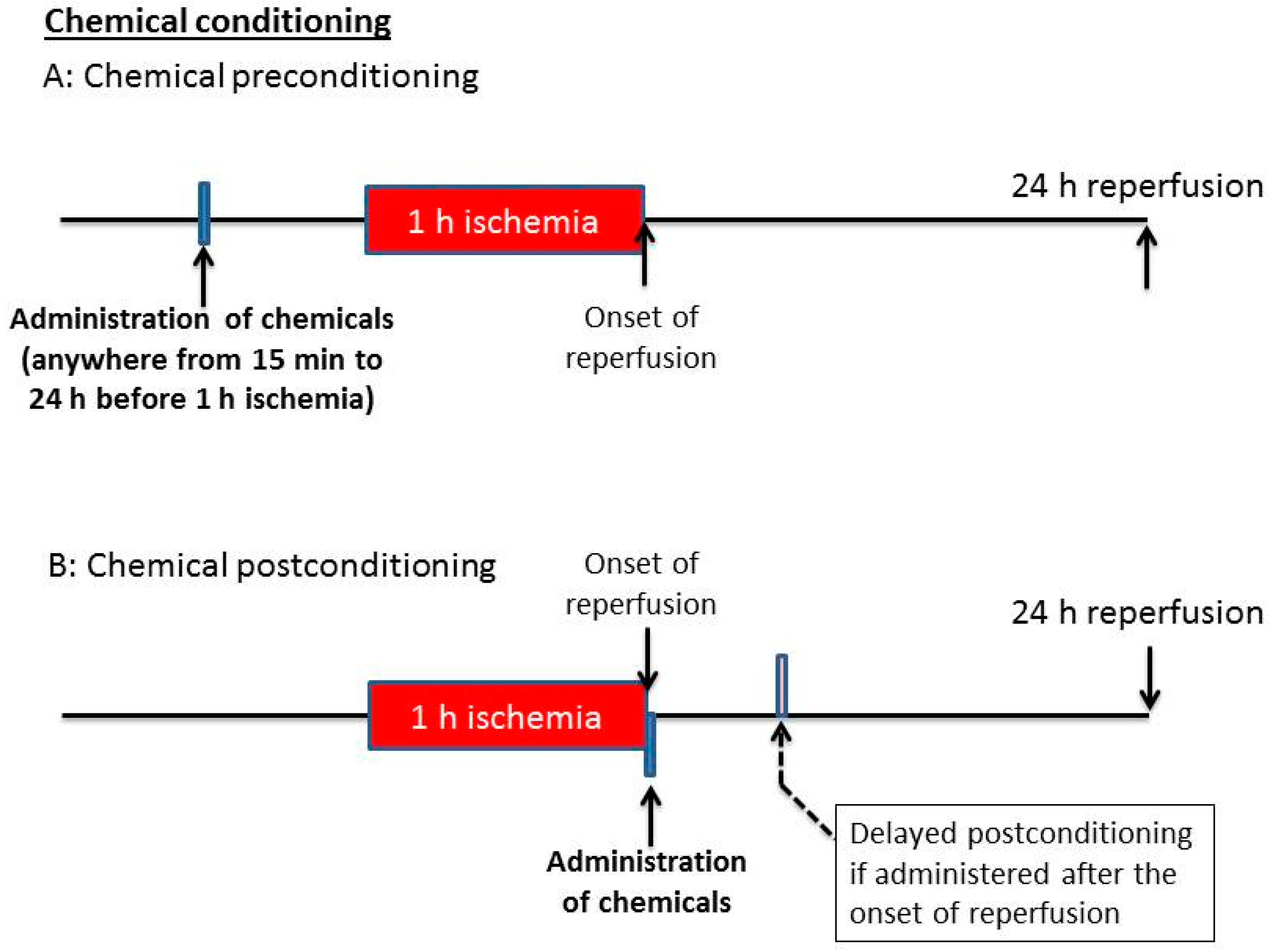
5. Chemical Conditioning: Chemical Pre-C and Post-C
6. Components of Mitochondrial Metabolic Pathways as Targets of Chemical Conditioning
6.1. Complex I Inhibition by Isoflurane
6.2. Inhibition of Succinate Dehydrogenase (Complex II) by 3-Nitropropionate
6.3. Preconditioning of Cytochrome c Oxidase (Complex IV) by Cyanide
6.4. Inhibition of Adenine Nucleotide Translocase by Carbon Monoxide
6.5. Inhibition of Mitochondrial Permeability Transition Pore by Carbon Dioxide
6.6. Activation of the Mitochondrial ATP-Sensitive K+ Channel by Diazoxide
6.7. Mitochondrial Biogenesis and Ischemic Tolerance
7. Summary and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kitagawa, K.; Matsumoto, M.; Tagaya, M.; Hata, R.; Ueda, H.; Niinobe, M.; Handa, N.; Fukunaga, R.; Kimura, K.; Mikoshiba, K.; et al. “Ischemic tolerance” phenomenon found in the brain. Brain Res. 1990, 528, 21–24. [Google Scholar] [CrossRef]
- Della-Morte, D.; Guadagni, F.; Palmirotta, R.; Ferroni, P.; Testa, G.; Cacciatore, F.; Abete, P.; Rengo, F.; Perez-Pinzon, M.A.; Sacco, R.L.; et al. Genetics and genomics of ischemic tolerance: Focus on cardiac and cerebral ischemic preconditioning. Pharmacogenomics 2012, 13, 1741–1757. [Google Scholar] [CrossRef] [PubMed]
- Prass, K.; Scharff, A.; Ruscher, K.; Lowl, D.; Muselmann, C.; Victorov, I.; Kapinya, K.; Dirnagl, U.; Meisel, A. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003, 34, 1981–1986. [Google Scholar] [CrossRef] [PubMed]
- Obrenovitch, T.P. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol. Rev. 2008, 88, 211–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Reis, C.; Applegate, R., 2nd; Stier, G.; Martin, R.; Zhang, J.H. Ischemic conditioning-induced endogenous brain protection: Applications pre-, per- or post-stroke. Exp. Neurol. 2015, 272, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, O.; Vinciguerra, A.; Cerullo, P.; Anzilotti, S.; Brancaccio, P.; Bilo, L.; Scorziello, A.; Molinaro, P.; di Renzo, G.; Pignataro, G. Ionic homeostasis in brain conditioning. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Li, X.; Peng, Y. Remote ischemic conditioning for acute ischemic stroke: Dawn in the darkness. Rev. Neurosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Abe, K. Recent progress in therapeutic strategy for ischemic stroke. Cell Transplant. 2016. [Google Scholar] [CrossRef]
- Fairbanks, S.L.; Brambrink, A.M. Preconditioning and postconditioning for neuroprotection: The most recent evidence. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, G.; Cuomo, O.; Vinciguerra, A.; Sirabella, R.; Esposito, E.; Boscia, F.; di Renzo, G.; Annunziato, L. Ncx as a key player in the neuroprotection exerted by ischemic preconditioning and postconditioning. Adv. Exp. Med. Biol. 2013, 961, 223–240. [Google Scholar] [PubMed]
- Pignataro, G.; Scorziello, A.; di Renzo, G.; Annunziato, L. Post-ischemic brain damage: Effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. Febs J. 2009, 276, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J. Cereb. Blood Flow Metab. 2009, 29, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Deguchi, K.; Sehara, Y.; Lukic-Panin, V.; Zhang, H.; Kamiya, T.; Abe, K. Therapeutic strategy for ischemic stroke. Neurochem. Res. 2009, 34, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Dezfulian, C.; Garrett, M.; Gonzalez, N.R. Clinical application of preconditioning and postconditioning to achieve neuroprotection. Transl. Stroke Res. 2013, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.; Samoilov, M. Current insights into the molecular mechanisms of hypoxic pre- and postconditioning using hypobaric hypoxia. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Desai, R.; Ji, X.; Lo, E.H. Pharmacologic pre- and postconditioning for stroke: Basic mechanisms and translational opportunity. Brain Circ. 2015, 1, 104–113. [Google Scholar]
- Sharma, A.; Goyal, R. Experimental brain ischemic preconditioning: A concept to putative targets. CNS Neurol. Disord. Drug Targets 2015, in press. [Google Scholar]
- Hassell, K.J.; Ezzati, M.; Alonso-Alconada, D.; Hausenloy, D.J.; Robertson, N.J. New horizons for newborn brain protection: Enhancing endogenous neuroprotection. Arch. Dis. Child. Fetal Neonatal. Ed. 2015, 100, F541–F552. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Becker, K.; Meisel, A. Preconditioning and tolerance against cerebral ischaemia: From experimental strategies to clinical use. Lancet Neurol. 2009, 8, 398–412. [Google Scholar] [CrossRef]
- Dirnagl, U.; Meisel, A. Endogenous neuroprotection: Mitochondria as gateways to cerebral preconditioning? Neuropharmacology 2008, 55, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.P.; Patel, P.M. Ischemic preconditioning in the brain. Curr. Opin. Anaesthesiol. 2003, 16, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.A.; Almeida, A.; Bolanos, J.P.; Lizasoain, I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic. Biol. Med. 2005, 39, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R.; Anderson, M.F. Mitochondrial contributions to tissue damage in stroke. Neurochem. Int. 2002, 40, 511–526. [Google Scholar] [CrossRef]
- Zhan, R.Z.; Fujihara, H.; Baba, H.; Yamakura, T.; Shimoji, K. Ischemic preconditioning is capable of inducing mitochondrial tolerance in the rat brain. Anesthesiology 2002, 97, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Papa, S.; Bolanos, J.; Bruckdorfer, R.; Carlsen, H.; Elliott, R.M.; Flier, J.; Griffiths, H.R.; Heales, S.; Holst, B.; et al. Antioxidants, reactive oxygen and nitrogen species, gene induction and mitochondrial function. Mol. Asp. Med. 2002, 23, 209–285. [Google Scholar] [CrossRef]
- Warner, D.S.; Sheng, H.; Batinic-Haberle, I. Oxidants, antioxidants and the ischemic brain. J. Exp. Biol. 2004, 207, 3221–3231. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol. 2012, 942, 93–136. [Google Scholar] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, atp, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Genova, M.L.; Pich, M.M.; Bernacchia, A.; Bianchi, C.; Biondi, A.; Bovina, C.; Falasca, A.I.; Formiggini, G.; Castelli, G.P.; Lenaz, G. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann. N. Y. Acad. Sci. 2004, 1011, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Halestrap, A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995, 307, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Luo, X.; Yan, L.J. Two dimensional blue native/SDS-PAGE to identify mitochondrial complex I subunits modified by 4-hydroxynonenal (HNE). Front. Physiol. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J. Analysis of oxidative modification of proteins. Curr. Protoc. Protein Sci. 2009. [Google Scholar] [CrossRef]
- Anderson, E.J.; Katunga, L.A.; Willis, M.S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 2012, 39, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Shigenaga, M.K. Oxidants are a major contributor to aging. Ann. N. Y. Acad. Sci. 1992, 663, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Shacter, E. Protein oxidative damage. Methods Enzymol. 2000, 319, 428–436. [Google Scholar] [PubMed]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Hoffman, D.L. Aging and cardiac ischemia-mitochondria and free radical consideration. In Oxidative Stress in Aging: From Model Systems to Human Diseases; Miwa, S., Bechkman, K.B., Muller, F.L., Eds.; Humana Press: Totowa, NJ, USA, 2008; pp. 253–268. [Google Scholar]
- Lebuffe, G.; Schumacker, P.T.; Shao, Z.H.; Anderson, T.; Iwase, H.; Vanden Hoek, T.L. ROS and NO trigger early preconditioning: Relationship to mitochondrial KATP channel. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H299–H308. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Xu, L.; Rozanski, D.J.; Sugawara, T.; Chan, P.H.; Trzaskos, J.M.; Feuerstein, G.Z. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: Exploration of potential mechanism associated with apoptosis. J. Pharmacol. Exp. Ther. 2003, 304, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.W.; Xia, Q.; Bruce, I.C. Reactive oxygen species mediate the neuroprotection conferred by a mitochondrial ATP-sensitive potassium channel opener during ischemia in the rat hippocampal slice. Brain Res. 2005, 1042, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Pong, K. Ischaemic preconditioning: Therapeutic implications for stroke? Expert Opin. Ther. Targets 2004, 8, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Lizasoain, I.; Sobrino, T.; Vivancos, J.; Castillo, J. Ischemic preconditioning: A novel target for neuroprotective therapy. Cerebrovasc. Dis. 2006, 21, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Kirino, T. Ischemic tolerance. J. Cereb. Blood Flow Metab. 2002, 22, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H. The protective effect of ischemic postconditioning against ischemic injury: From the heart to the brain. J. Neuroimmune Pharmacol. 2007, 2, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.W.; Dave, K.R.; Young, J.I.; Perez-Pinzon, M.A. Ischemic preconditioning alters the epigenetic profile of the brain from ischemic intolerance to ischemic tolerance. Neurotherapeutics 2013, 10, 789–797. [Google Scholar] [CrossRef] [PubMed]
- N, T.V.; Sangwan, A.; Sharma, B.; Majid, A.; Gk, R. Cerebral ischemic preconditioning: The road so far. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ren, C.; Chen, X.; Shen, J. From rapid to delayed and remote postconditioning: The evolving concept of ischemic postconditioning in brain ischemia. Curr. Drug Targets 2012, 13, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Stowe, A.M.; Wacker, B.K.; Cravens, P.D.; Perfater, J.L.; Li, M.K.; Hu, R.; Freie, A.B.; Stuve, O.; Gidday, J.M. CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J. Neuroinflamm. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Galle, A.A.; Jones, N.M. The neuroprotective actions of hypoxic preconditioning and postconditioning in a neonatal rat model of hypoxic-ischemic brain injury. Brain Res. 2013, 1498, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Stowe, A.M.; Altay, T.; Freie, A.B.; Gidday, J.M. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann. Neurol. 2011, 69, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Riepe, M.W.; Ludolph, A.C. Chemical preconditioning: A cytoprotective strategy. Mol. Cell. Biochem. 1997, 174, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, A.; Nakahara, T.; Kayama, H.; Yamamoto, T. Ischemic tolerance in chemical preconditioning: Possible role of astrocytic glutamine synthetase buffering glutamate-mediated neurotoxicity. J. Neurosci. Res. 2006, 84, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Riepe, M.W.; Esclaire, F.; Kasischke, K.; Schreiber, S.; Nakase, H.; Kempski, O.; Ludolph, A.C.; Dirnagl, U.; Hugon, J. Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: “Chemical preconditioning”. J. Cereb. Blood Flow Metab. 1997, 17, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Leak, R.K.; Gan, Y.; Li, P.; Zhang, F.; Hu, X.; Jing, Z.; Chen, J.; Zigmond, M.J.; Gao, Y. Preconditioning provides neuroprotection in models of CNS disease: Paradigms and clinical significance. Prog. Neurobiol. 2014, 114, 58–83. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.I.; Lee, Y.W.; Kim, Y.K. Chemical hypoxia-induced cell death in human glioma cells: Role of reactive oxygen species, ATP depletion, mitochondrial damage and Ca2+. Neurochem. Res. 2003, 28, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Roemgens, A.; Singh, S.; Beyer, C.; Arnold, S. Inducers of chemical hypoxia act in a gender- and brain region-specific manner on primary astrocyte viability and cytochrome c oxidase. Neurotox. Res. 2011, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Danielisova, V.; Gottlieb, M.; Nemethova, M.; Kravcukova, P.; Domorakova, I.; Mechirova, E.; Burda, J. Bradykinin postconditioning protects pyramidal Ca1 neurons against delayed neuronal death in rat hippocampus. Cell. Mol. Neurobiol. 2009, 29, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, R.; Yan, L.J. Roles of pyruvate, nadh, and mitochondrial complex I in redox balance and imbalance in β cell function and dysfunction. J. Diabetes Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wu, J.; Jing, S.; Yan, L.J. Hyperglycemic stress and carbon stress in diabetic glucotoxicity. Aging Dis. 2016, 7, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, M.; Marks, A.D. Marks’ Basic Medical Biochemistry: A Clinical Approach, 4th ed.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Sun, X.; Sun, J.; Liu, L.; Ding, Z. Isoflurane preconditioning and postconditioning in multiple organ protection. J. Biochem. Pharmacol. Res. 2013, 1, 6–14. [Google Scholar]
- Hanley, P.J.; Ray, J.; Brandt, U.; Daut, J. Halothane, isoflurane and sevoflurane inhibit NADH: Ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J. Physiol. 2002, 544, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Kayser, E.B.; Suthammarak, W.; Morgan, P.G.; Sedensky, M.M. Isoflurane selectively inhibits distal mitochondrial complex I in Caenorhabditis elegans. Anesth. Analg. 2011, 112, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Pravdic, D.; Hirata, N.; Barber, L.; Sedlic, F.; Bosnjak, Z.J.; Bienengraeber, M. Complex I and ATP synthase mediate membrane depolarization and matrix acidification by isoflurane in mitochondria. Eur. J. Pharmacol. 2012, 690, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, B.; Dash, R.K.; Stowe, D.F.; Bosnjak, Z.J.; Camara, A.K. Isoflurane modulates cardiac mitochondrial bioenergetics by selectively attenuating respiratory complexes. Biochim. Biophys. Acta 2014, 1837, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Sosunov, S.A.; Ameer, X.; Niatsetskaya, Z.V.; Utkina-Sosunova, I.; Ratner, V.I.; Ten, V.S. Isoflurane anesthesia initiated at the onset of reperfusion attenuates oxidative and hypoxic-ischemic brain injury. PLoS ONE 2015, 10, e0120456. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Human complex I defects in neurodegenerative diseases. Biochim. Biophys. Acta 1998, 1364, 261–270. [Google Scholar] [CrossRef]
- Lefort, N.; Glancy, B.; Bowen, B.; Willis, W.T.; Bailowitz, Z.; de Filippis, E.A.; Brophy, C.; Meyer, C.; Hojlund, K.; Yi, Z.; et al. Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes 2010, 59, 2444–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex i. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, L.M.; Tanaka, K.; Eells, J.T.; Weihrauch, D.; Pagel, P.S.; Kersten, J.R.; Warltier, D.C. Preconditioning by isoflurane is mediated by reactive oxygen species generated from mitochondrial electron transport chain complex III. Anesth. Analg. 2004, 99, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.; Chiari, P.; Fauconnier, J.; Abrial, M.; Couture-Lepetit, E.; Harisseh, R.; Pillot, B.; Lacampagne, A.; Tourneur, Y.; Gharib, A.; et al. Involvement of cyclophilin D and calcium in isoflurane-induced preconditioning. Anesthesiology 2015, 123, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Ji, G.; Xue, H.; Yu, W.; Zhao, X.; Ding, M.; Yang, Y.; Zuo, Z. Isoflurane postconditioning improved long-term neurological outcome possibly via inhibiting the mitochondrial permeability transition pore in neonatal rats after brain hypoxia-ischemia. Neuroscience 2014, 280, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Bove, J.; Vila, M.; Przedborski, S. The rotenone model of parkinson’s disease. Trends Neurosci. 2003, 26, 345–346. [Google Scholar] [CrossRef]
- Schonfeld, P.; Reiser, G. Rotenone-like action of the branched-chain phytanic acid induces oxidative stress in mitochondria. J. Biol. Chem. 2006, 281, 7136–7142. [Google Scholar] [CrossRef] [PubMed]
- Marella, M.; Seo, B.B.; Nakamaru-Ogiso, E.; Greenamyre, J.T.; Matsuno-Yagi, A.; Yagi, T. Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of parkinson’s disease. PLoS ONE 2008, 3, e1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Liu, C.; Liu, W.; Zhang, H.; Zhang, R.; Liu, J.; Zhang, J.; Xu, C.; Liu, L.; Huang, S.; et al. Rotenone induction of hydrogen peroxide inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E pathways, leading to neuronal apoptosis. Toxicol. Sci. 2015, 143, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Sumien, N.; Thangthaeng, N.; Forster, M.J. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic. Res. 2013, 47, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Fukuse, T.; Ishikawa, S.; Hanaoka, S.; Chen, Q.; Shoji, T.; Wada, H. “Chemical preconditioning” by 3-nitropropionate reduces ischemia-reperfusion injury in cardiac-arrested rat lungs. Transplantation 2001, 71, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Brambrink, A.M.; Schneider, A.; Noga, H.; Astheimer, A.; Gotz, B.; Korner, I.; Heimann, A.; Welschof, M.; Kempski, O. Tolerance-inducing dose of 3-nitropropionic acid modulates bcl-2 and bax balance in the rat brain: A potential mechanism of chemical preconditioning. J. Cereb. Blood Flow Metab. 2000, 20, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, A.; Nakahara, T.; Ogata, M.; Yamamoto, T. The critical threshold of 3-nitropropionic acid-induced ischemic tolerance in the rat. Brain Res. 2005, 1050, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, F.; Liao, W.; Busch, C.; Castell, S.; Knapp, F.; Lindauer, U.; Megow, D.; Meisel, A.; Redetzky, A.; Ruscher, K.; et al. Respiratory chain inhibition induces tolerance to focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1999, 19, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E.; Brown, G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Santos, R.X.; Cardoso, S.M.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Cyanide preconditioning protects brain endothelial and NT2 neuron-like cells against glucotoxicity: Role of mitochondrial reactive oxygen species and HIF-1α. Neurobiol. Dis. 2012, 45, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.S.; Ahlemeyer, B.; Ravati, A.; Thakur, P.; Mennel, H.D.; Krieglstein, J. Preconditioning-induced protection against cyanide-induced neurotoxicity is mediated by preserving mitochondrial function. Neurochem. Int. 2002, 40, 285–293. [Google Scholar] [CrossRef]
- Queiroga, C.S.; Almeida, A.S.; Martel, C.; Brenner, C.; Alves, P.M.; Vieira, H.L. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J. Biol. Chem. 2010, 285, 17077–17088. [Google Scholar] [CrossRef] [PubMed]
- Hochgrafe, F.; Mostertz, J.; Albrecht, D.; Hecker, M. Fluorescence thiol modification assay: Oxidatively modified proteins in Bacillus subtilis. Mol. Microbiol. 2005, 58, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Davidson, S.M.; Hausenloy, D.J.; Yellon, D.M. Preconditioning and postconditioning: The essential role of the mitochondrial permeability transition pore. Cardiovasc. Res. 2007, 75, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Christians, E.S.; Liu, L.; Xiao, X.; Sohal, R.S.; Benjamin, I.J. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002, 21, 5164–5172. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Y.; Shen, Z.; He, P.; Jiang, L.; Hou, W.W.; Shen, Y.; Zhang, X.N.; Hu, W.W.; Chen, Z. A novel neuroprotective strategy for ischemic stroke: Transient mild acidosis treatment by CO2 inhalation at reperfusion. J. Cereb. Blood Flow Metab. 2014, 34, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Garcia de Arriba, S.; Franke, H.; Pissarek, M.; Nieber, K.; Illes, P. Neuroprotection by ATP-dependent potassium channels in rat neocortical brain slices during hypoxia. Neurosci. Lett. 1999, 273, 13–16. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kashii, S.; Yasuyoshi, H.; Zhang, S.; Honda, Y.; Akaike, A. Mitochondrial ATP-sensitive potassium channel: A novel site for neuroprotection. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2750–2756. [Google Scholar] [CrossRef]
- Kis, B.; Rajapakse, N.C.; Snipes, J.A.; Nagy, K.; Horiguchi, T.; Busija, D.W. Diazoxide induces delayed pre-conditioning in cultured rat cortical neurons. J. Neurochem. 2003, 87, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Robin, E.; Simerabet, M.; Hassoun, S.M.; Adamczyk, S.; Tavernier, B.; Vallet, B.; Bordet, R.; Lebuffe, G. Postconditioning in focal cerebral ischemia: Role of the mitochondrial ATP-dependent potassium channel. Brain Res. 2011, 1375, 137–146. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.C.; Yao, X.L.; Alam, H.; McCabe, J.T. Diazoxide, as a postconditioning and delayed preconditioning trigger, increases HSP25 and HSP70 in the central nervous system following combined cerebral stroke and hemorrhagic shock. J. Neurotrauma 2007, 24, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Shen, F.; Lin, L.; Zhang, X.; Bruce, I.C.; Xia, Q. The neuroprotection conferred by activating the mitochondrial ATP-sensitive K+ channel is mediated by inhibiting the mitochondrial permeability transition pore. Neurosci. Lett. 2006, 402, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Timmer, N.M.; Domoki, F.; Mihaly, A.; Luiten, P.G.; Bari, F. Post-ischemic administration of diazoxide attenuates long-term microglial activation in the rat brain after permanent carotid artery occlusion. Neurosci. Lett. 2005, 387, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Leak, R.K.; Yin, W.; Zhang, L.; Wang, S.; Gao, Y.; Chen, J. Mitochondrial biogenesis contributes to ischemic neuroprotection afforded by LPS pre-conditioning. J. Neurochem. 2012, 123, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, H.L.; Minami, M.; Lessov, N.S.; Coste, S.C.; Stevens, S.L.; Henshall, D.C.; Meller, R.; Simon, R.P.; Stenzel-Poore, M.P. Endotoxin preconditioning protects against the cytotoxic effects of TNFα after stroke: A novel role for TNFα in LPS-ischemic tolerance. J. Cereb. Blood Flow Metab. 2007, 27, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, K.B.; Stevens, S.L.; Marsh, B.J.; Williams-Karnesky, R.; Lessov, N.S.; Stenzel-Poore, M.P. LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J. Neuroinflamm. 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Marsh, B.; Stevens, S.L.; Packard, A.E.; Gopalan, B.; Hunter, B.; Leung, P.Y.; Harrington, C.A.; Stenzel-Poore, M.P. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: A critical role for IRF3. J. Neurosci. 2009, 29, 9839–9849. [Google Scholar] [CrossRef] [PubMed]
- Li, W.C.; Jiang, R.; Jiang, D.M.; Zhu, F.C.; Su, B.; Qiao, B.; Qi, X.T. Lipopolysaccharide preconditioning attenuates apoptotic processes and improves neuropathologic changes after spinal cord injury in rats. Int. J. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Z.; Wu, J.; Yan, L.-J. Chemical Conditioning as an Approach to Ischemic Stroke Tolerance: Mitochondria as the Target. Int. J. Mol. Sci. 2016, 17, 351. https://doi.org/10.3390/ijms17030351
Jin Z, Wu J, Yan L-J. Chemical Conditioning as an Approach to Ischemic Stroke Tolerance: Mitochondria as the Target. International Journal of Molecular Sciences. 2016; 17(3):351. https://doi.org/10.3390/ijms17030351
Chicago/Turabian StyleJin, Zhen, Jinzi Wu, and Liang-Jun Yan. 2016. "Chemical Conditioning as an Approach to Ischemic Stroke Tolerance: Mitochondria as the Target" International Journal of Molecular Sciences 17, no. 3: 351. https://doi.org/10.3390/ijms17030351