
Autophagic Cell Death and Apoptosis Jointly Mediate Cisatracurium Besylate-Induced Cell Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
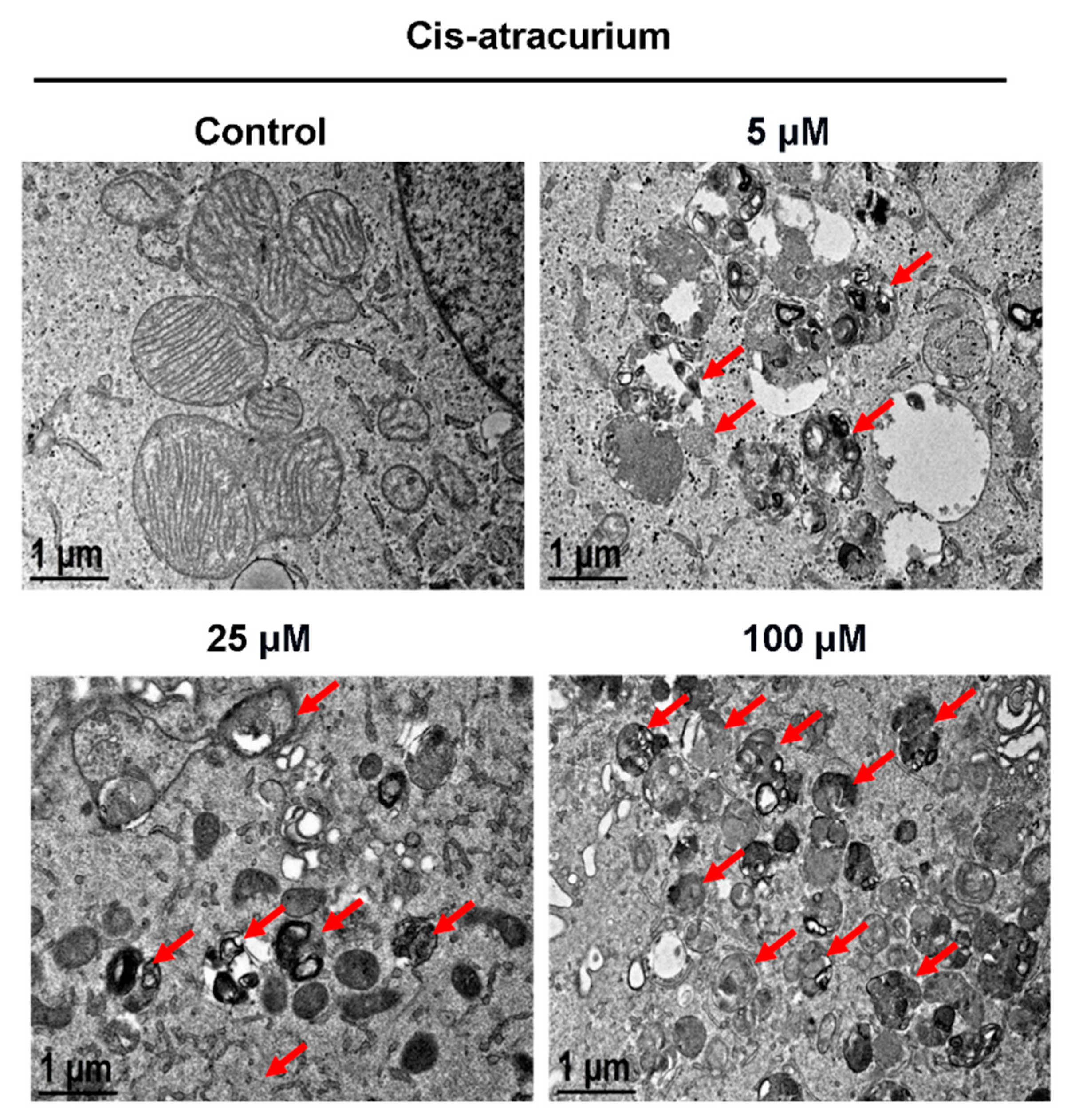
2.1. Cisatracurium Besylate Promotes Mitochondrial Breakage, Mitochondrial Protein Degradation and Colocalization of Mitochondria and Autophagosomes
2.2. Cisatracurium Besylate Promotes Autolysosome Formation
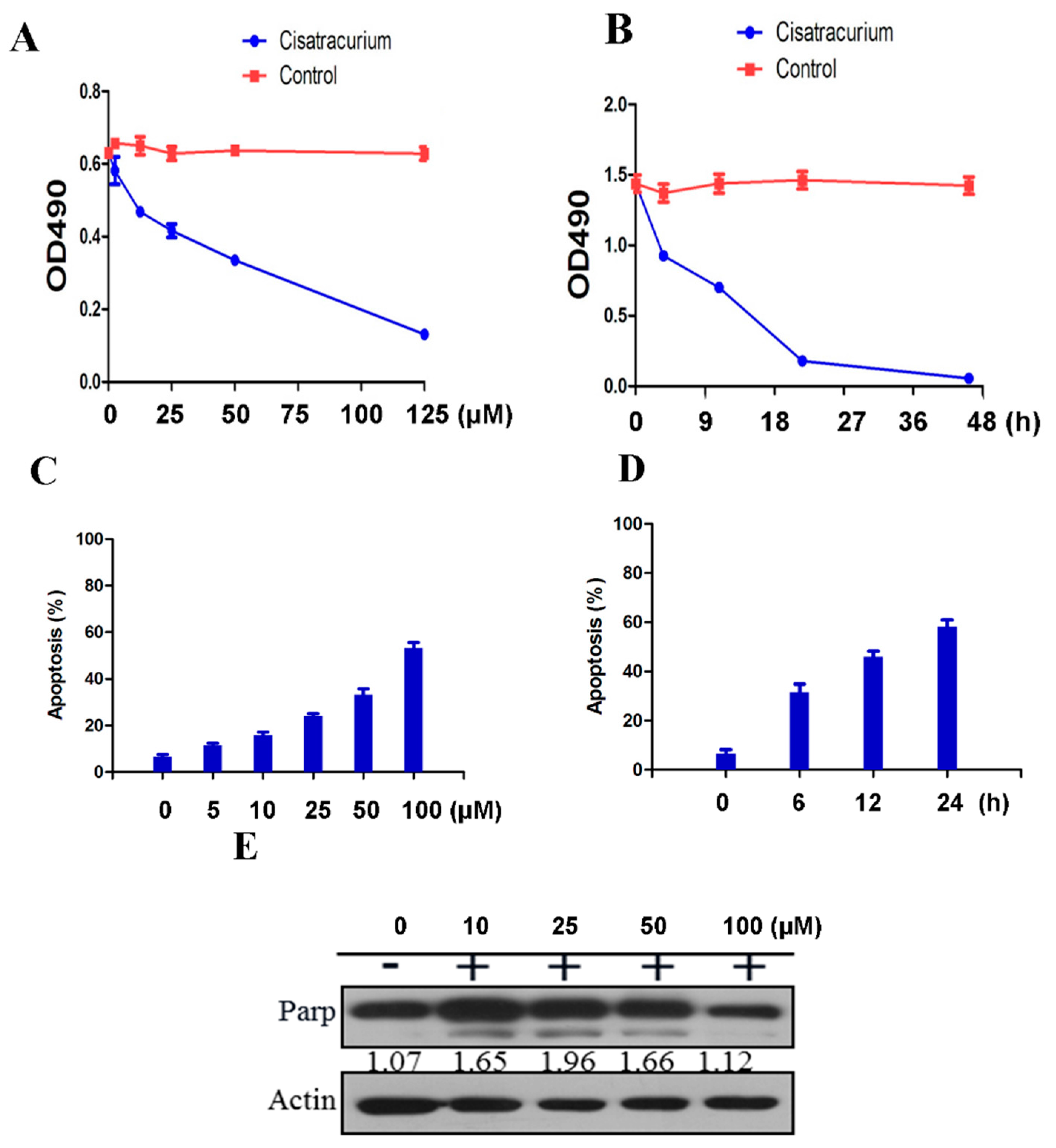
2.3. Cisatracurium Besylate Inhibits Proliferation of Endothelial Cells in Vitro
2.4. Cisatracurium Besylate Promotes Apoptosis of Endothelial Cells in Vitro
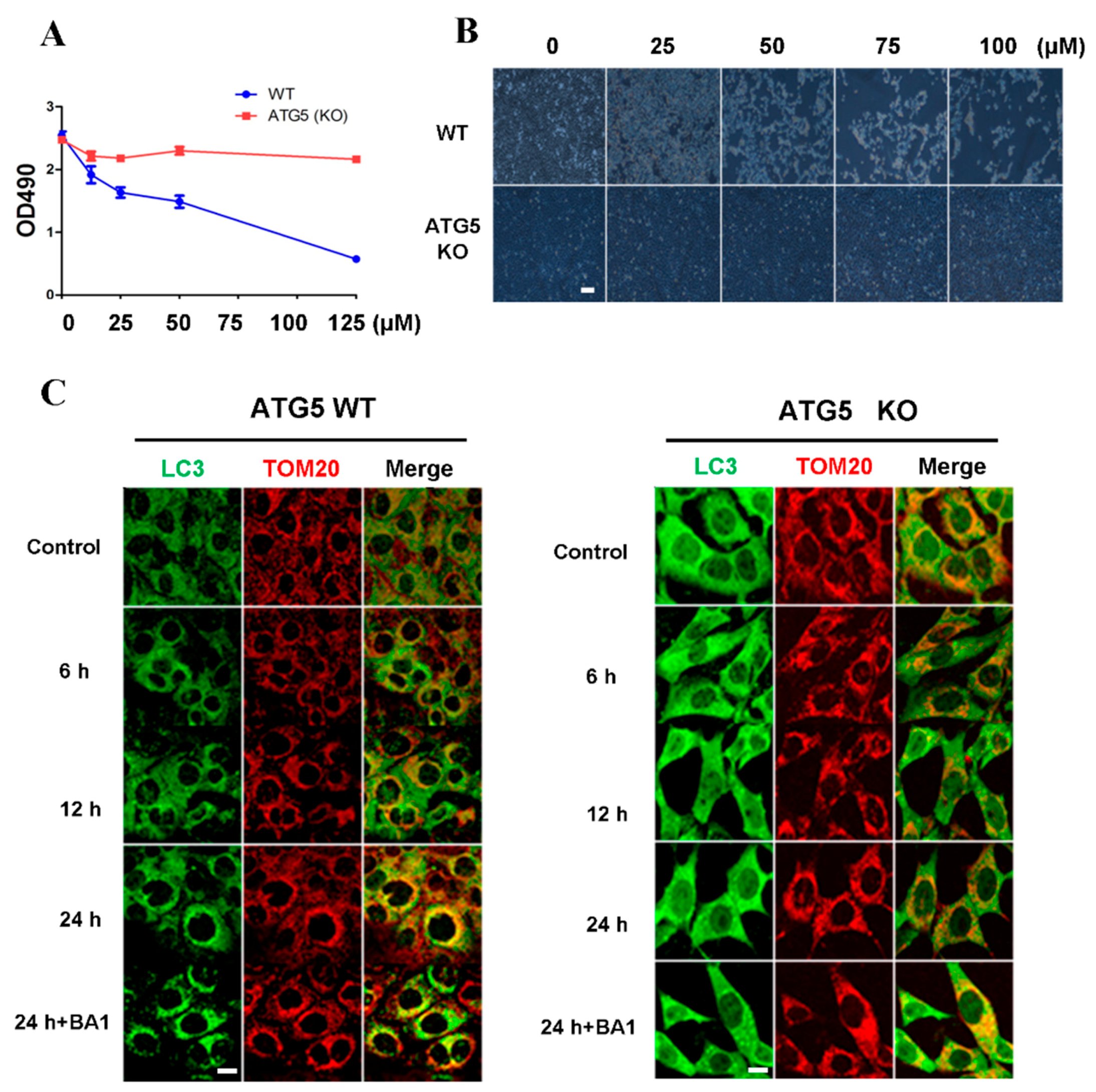
2.5. Cell Death Induced by Cisatracurium Besylate Depends on Autophagy
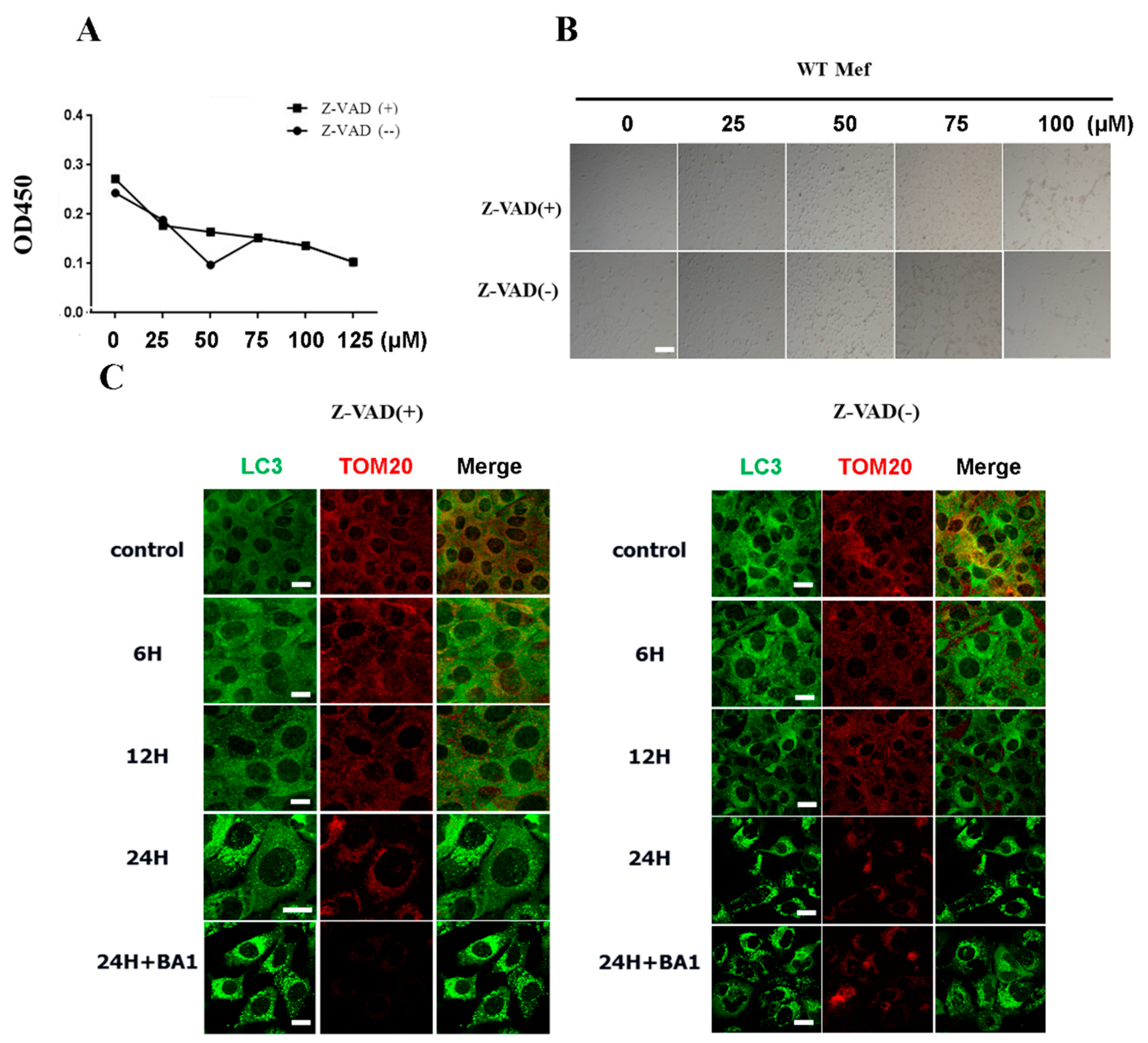
2.6. Cisatracurium Besylate Can Promote Cell Death Even in the Presence of the Apoptosis Inhibitor zVAD
3. Discussion
4. Experimental Section
4.1. Cell Culture and Antibodies
4.2. Cell Viability Assay
4.3. Cell Apoptosis Assay
4.4. Western Blot Analysis
4.5. Immunofluorescence Microscopy
4.6. Electron Microscopy
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Wolf, A.M.; Freeman, J.A.; Scott, V.L.; Tullock, W.; Smith, D.A.; Kisor, D.F.; Kerls, S.; Cook, D.R. Pharmacokinetics and pharmacodynamics of cisatracurium in patients with end-stage liver disease undergoing liver transplantation. Br. J. Anaesth. 1996, 76, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Jirasiritham, S.; Tantivitayatan, K. A comparison of the efficacy of cisatracurium and atracurium in kidney transplantation operation. J. Med. Assoc. Thai 2004, 87, 73–79. [Google Scholar] [PubMed]
- De Rossi, L.; Fritz, H.; Klein, U. Comparison of cisatracurium-induced neuromuscular block at the masseter and adductor pollicis muscle. Eur. J. Anaesthesiol. 2000, 17, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Serra, C.S.; Oliveira, A.C. Cisatracurium: Myographical and electrophysiological studies in the isolated rat muscle. Fundam. Clin. Pharmacol. 2006, 20, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, C.; Borrallo, J.M.; Romera, M.A.; Silva, J.A.; Balandin, B. Anesthesia and analgesia protocol during therapeutic hypothermia after cardiac arrest: A systematic review. Anesth. Analg. 2010, 110, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yamaguchi, N.; Varin, F. Dose-dependency of pharmacokinetic/pharmacodynamic parameters after intravenous bolus doses of cisatracurium. Br. J. Anaesth. 2008, 101, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Amann, A.; Rieder, J.; Fleischer, M.; Niedermuller, P.; Hoffmann, G.; Amberger, A.; Marth, C.; Nigrovic, V.; Puhringer, F. The influence of atracurium, cisatracurium, and mivacurium on the proliferation of two human cell lines in vitro. Anesth. Analg. 2001, 93, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Rieder, J.; Lirk, P.; Bodrogi, F.; Sawires, M.; Gruber, G.; Hoffmann, G. Cisatracurium, but not mivacurium, induces apoptosis in human umbilical vein endothelial cells in vitro. Eur. J. Anaesthesiol. 2005, 22, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Rubinsztein, D.C. Atg5 and Bcl-2 provide novel insights into the interplay between apoptosis and autophagy. Cell Death Differ. 2007, 14, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human Atg12~Atg5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Li, W. Impairment of autophagosome-lysosome fusion in the buff mutant mice with the VPS33A mutation. Autophagy 2015, 11, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Klionsky, D.J. Toward an understanding of autophagosome-lysosome fusion: The unsuspected role of Atg14. Autophagy 2015, 11, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Puthalakath, H. Bcl-2 family proteins: The sentinels of the mitochondrial apoptosis pathway. IUBMB Life 2008, 60, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- El-Khattouti, A.; Selimovic, D.; Haikel, Y.; Hassan, M. Crosstalk between apoptosis and autophagy: Molecular mechanisms and therapeutic strategies in cancer. J. Cell Death 2013, 6, 37–55. [Google Scholar] [PubMed]
- Harr, M.W.; Distelhorst, C.W. Apoptosis and autophagy: Decoding calcium signals that mediate life or death. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Yamaguchi, O.; Otsu, K. Crosstalk between autophagy and apoptosis in heart disease. Circ. Res. 2008, 103, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Liu, L.; Zhu, Y.; Chen, Q. Molecular signaling toward mitophagy and its physiological significance. Exp. Cell Res. 2013, 319, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Matsui, M.; Mizushima, N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: Caution in the interpretation of LC3 localization. Autophagy 2007, 3, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Bampton, E.T.; Goemans, C.G.; Niranjan, D.; Mizushima, N.; Tolkovsky, A.M. The dynamics of autophagy visualized in live cells: From autophagosome formation to fusion with endo/lysosomes. Autophagy 2005, 1, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Niikura, K. Vacuolar ATPase as a drug discovery target. Drug News Perspect. 2006, 19, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, Y.; Futai, M.; Yoshimori, T.; Yamamoto, A.; Tashiro, Y. Bafilomycins and related compounds as vacuolar H(+)-atpase inhibitors (in Japanese). Tanpakushitsu Kakusan Koso 1993, 38, 2000–2011. [Google Scholar] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast APG8P, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Slobodkin, M.R.; Elazar, Z. The Atg8 family: Multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013, 55, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Shvets, E.; Elazar, Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 2011, 80, 125–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004, 36, 2445–2462. [Google Scholar] [CrossRef] [PubMed]
- Dotti, G.; Savoldo, B.; Pule, M.; Straathof, K.C.; Biagi, E.; Yvon, E.; Vigouroux, S.; Brenner, M.K.; Rooney, C.M. Human cytotoxic t lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood 2005, 105, 4677–4684. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Caspase activation, inhibition, and reactivation: A mechanistic view. Protein Sci. 2004, 13, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Sane, A.T.; Steyaert, A.; Cimoli, G.; Bertrand, R. The Bcl-xL and Bax-alpha control points: Modulation of apoptosis induced by cancer chemotherapy and relation to TPCK-sensitive protease and caspase activation. Biochem. Cell Biol. 1997, 75, 301–314. [Google Scholar] [PubMed]
- Walczak, M.; Martens, S. Dissecting the role of the Atg12–Atg5-Atg16 complex during autophagosome formation. Autophagy 2013, 9, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Feldman, T.; Jiang, X. Stimulation of Atg12–Atg5 conjugation by ribonucleic acid. Autophagy 2007, 3, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, J.; Miura, E.; Mizushima, N.; Watanabe, M.; Yuzaki, M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null purkinje cells before neuronal death. Autophagy 2007, 3, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Kimchi, A. Autophagy and cell death. Curr. Top. Dev. Biol. 2007, 78, 217–245. [Google Scholar] [PubMed]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an Atg7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Bursch, W.; Ellinger, A.; Gerner, C.; Frohwein, U.; Schulte-Hermann, R. Programmed cell death (PCD). Apoptosis, autophagic pcd, or others? Ann. N. Y. Acad. Sci. 2000, 926, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sator-Katzenschlager, S.M.; Oehmke, M.J.; Kontaratos, M.; Wedrich, A.; Heinze, G.; Weinstabl, C. Effect of different doses of cisatracurium on intraocular pressure in sedated patients. Eur. J. Anaesthesiol. 2002, 19, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Dhonneur, G.; Cerf, C.; Lagneau, F.; Mantz, J.; Gillotin, C.; Duvaldestin, P. The pharmacokinetics of cisatracurium in patients with acute respiratory distress syndrome. Anesth. Analg. 2001, 93, 400–404. [Google Scholar] [PubMed]
- Smith, C.E.; van Miert, M.M.; Parker, C.J.; Hunter, J.M. A comparison of the infusion pharmacokinetics and pharmacodynamics of cisatracurium, the 1R-cis 1’R-cis isomer of atracurium, with atracurium besylate in healthy patients. Anaesthesia 1997, 52, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Schramm, W.M.; Papousek, A.; Michalek-Sauberer, A.; Czech, T.; Illievich, U. The cerebral and cardiovascular effects of cisatracurium and atracurium in neurosurgical patients. Anesth. Analg. 1998, 86, 123–127. [Google Scholar] [PubMed]
- Lepage, J.Y.; Malinovsky, J.M.; Malinge, M.; Lechevalier, T.; Dupuch, C.; Cozian, A.; Pinaud, M.; Souron, R. Pharmacodynamic dose-response and safety study of cisatracurium (51W89) in adult surgical patients during N2O-O2-opioid anesthesia. Anesth. Analg. 1996, 83, 823–829. [Google Scholar] [PubMed]
- Sorooshian, S.S.; Stafford, M.A.; Eastwood, N.B.; Boyd, A.H.; Hull, C.J.; Wright, P.M. Pharmacokinetics and pharmacodynamics of cisatracurium in young and elderly adult patients. Anesthesiology 1996, 84, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Brauns, S.C.; Dealtry, G.; Milne, P.; Naude, R.; van de Venter, M. Caspase-3 activation and induction of PARP cleavage by cyclic dipeptide cyclo(Phe-Pro) in HT-29 cells. Anticancer Res. 2005, 25, 4197–4202. [Google Scholar] [PubMed]
- Decker, P.; Isenberg, D.; Muller, S. Inhibition of caspase-3-mediated poly(ADP-ribose) polymerase (PARP) apoptotic cleavage by human PARP autoantibodies and effect on cells undergoing apoptosis. J. Biol. Chem. 2000, 275, 9043–9046. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Li, W.; Chen, Y.; Yan, Z.; Huang, X.; Zhuang, H.; Zhong, W.; Wu, W.; Lin, C.; Chen, H.; et al. Phosphorylation of ulk1 by AMPK regulates translocation of ulk1 to mitochondria and mitophagy. FEBS Lett. 2015, 589, 1847–1854. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuang, H.; Tian, W.; Li, W.; Zhang, X.; Wang, J.; Yang, Y.; Liu, X.; Xia, Z.; Feng, D.; Zhang, L. Autophagic Cell Death and Apoptosis Jointly Mediate Cisatracurium Besylate-Induced Cell Injury. Int. J. Mol. Sci. 2016, 17, 515. https://doi.org/10.3390/ijms17040515
Zhuang H, Tian W, Li W, Zhang X, Wang J, Yang Y, Liu X, Xia Z, Feng D, Zhang L. Autophagic Cell Death and Apoptosis Jointly Mediate Cisatracurium Besylate-Induced Cell Injury. International Journal of Molecular Sciences. 2016; 17(4):515. https://doi.org/10.3390/ijms17040515
Chicago/Turabian StyleZhuang, Haixia, Weili Tian, Wen Li, Xingli Zhang, Jingjing Wang, Yue Yang, Xin Liu, Zhengyuan Xia, Du Feng, and Liangqing Zhang. 2016. "Autophagic Cell Death and Apoptosis Jointly Mediate Cisatracurium Besylate-Induced Cell Injury" International Journal of Molecular Sciences 17, no. 4: 515. https://doi.org/10.3390/ijms17040515