
Structural Investigation for Optimization of Anthranilic Acid Derivatives as Partial FXR Agonists by in Silico Approaches

Abstract
:1. Introduction
2. Results and Discussion
2.1. Two-Dimensional Quantitative Structure Activity Relationship (2D-QSAR) Models
2.1.1. Multiple Linear Regression Modeling
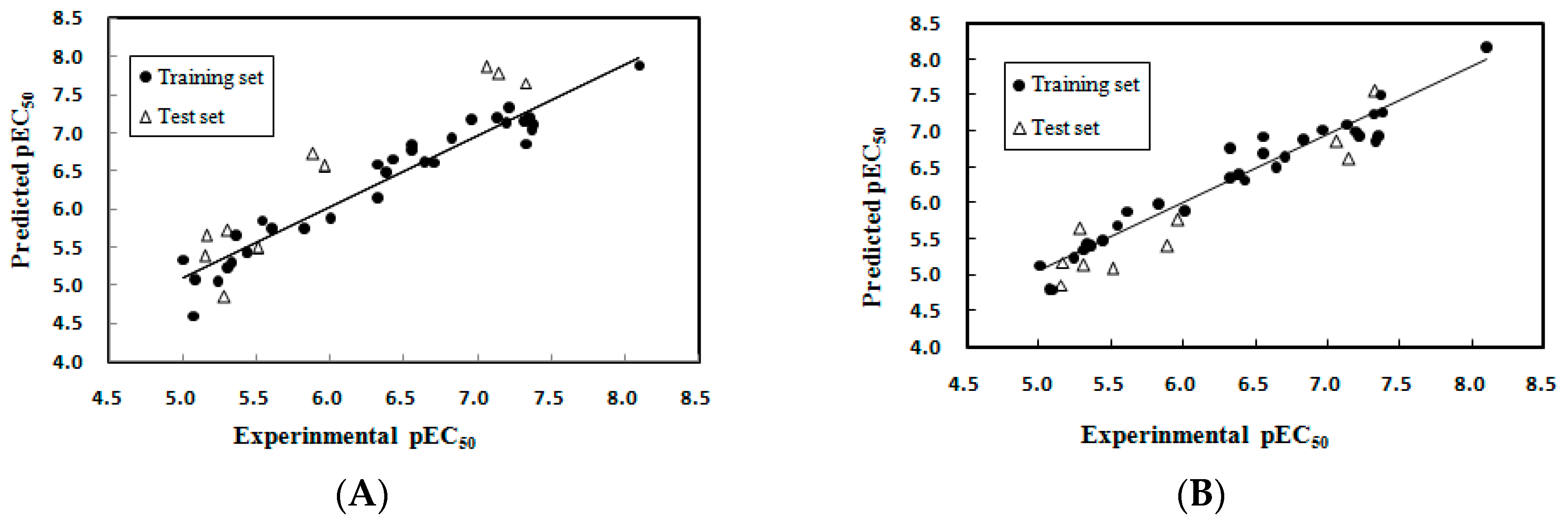
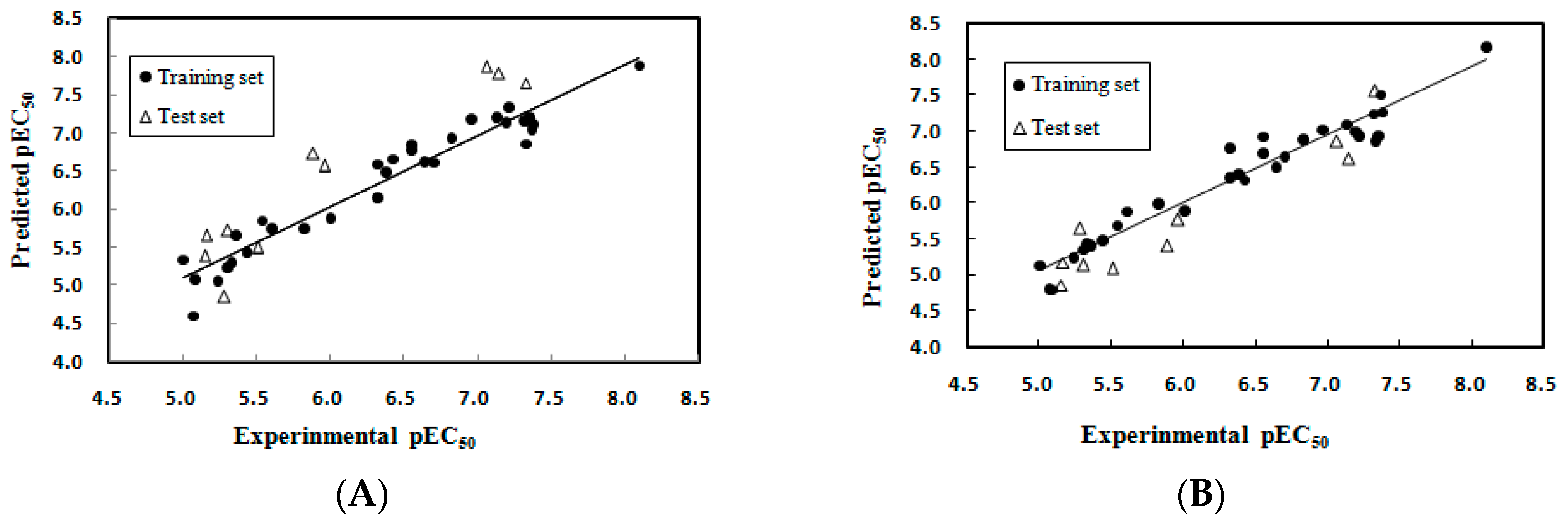
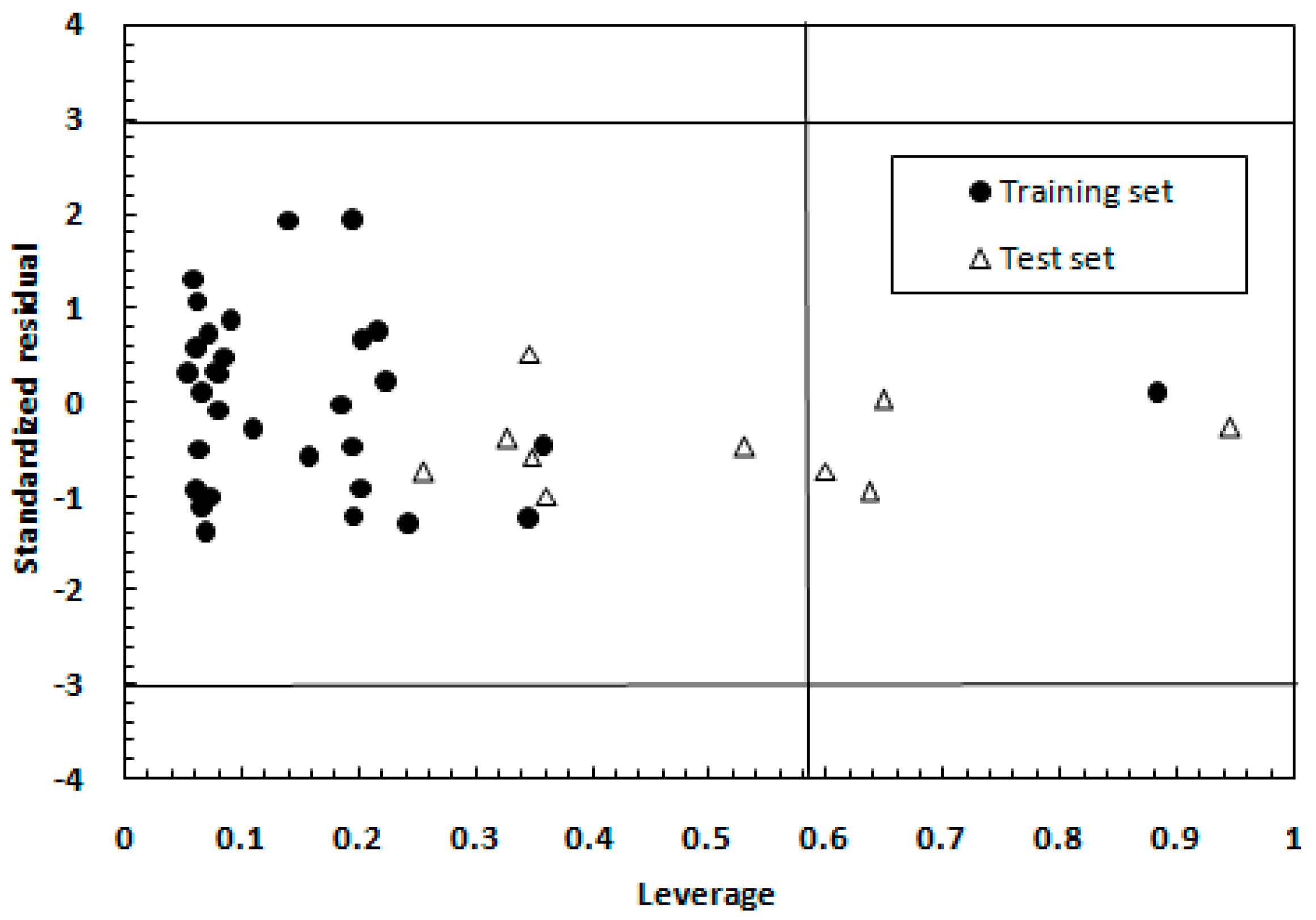
2.1.2. Model Applicability Domain Analysis for the MLR Model
2.1.3. Interpretation of the Descriptors
2.2. 3D-QSAR Models
2.2.1. CoMFA Analysis
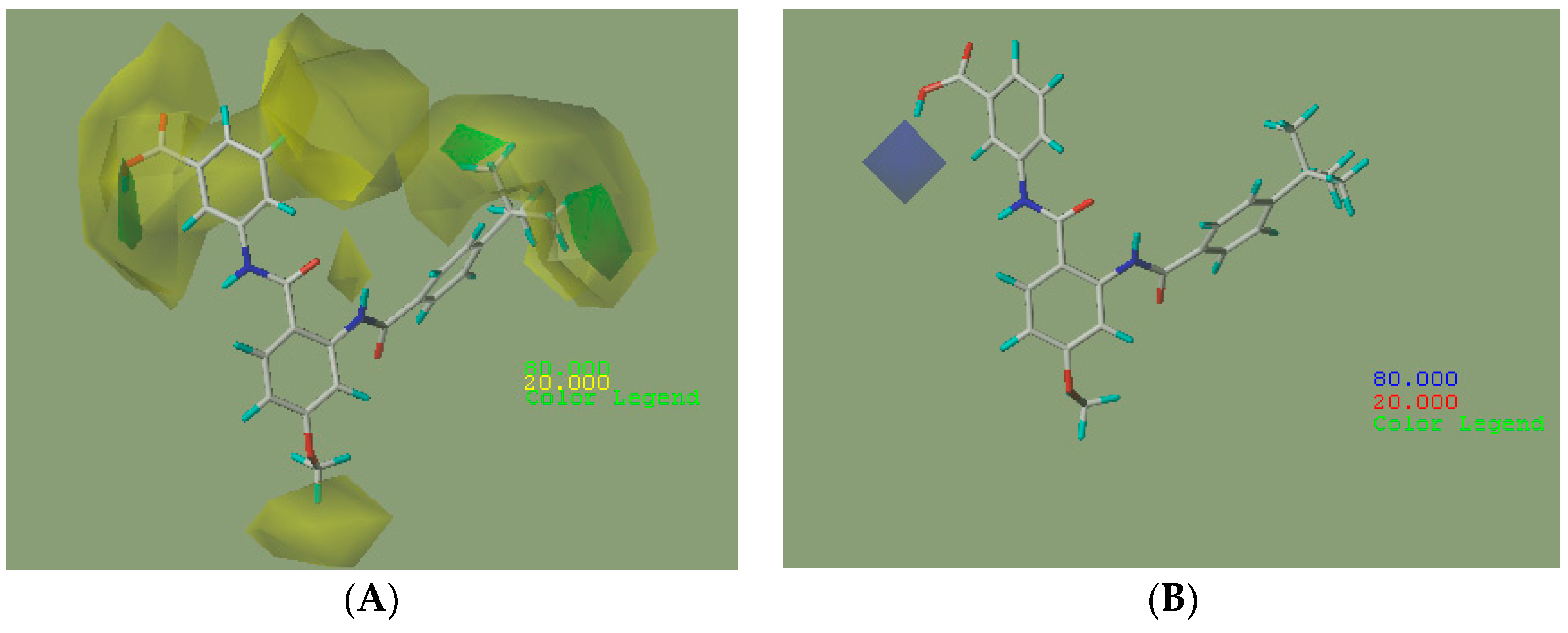
2.2.2. CoMFA Contour Maps
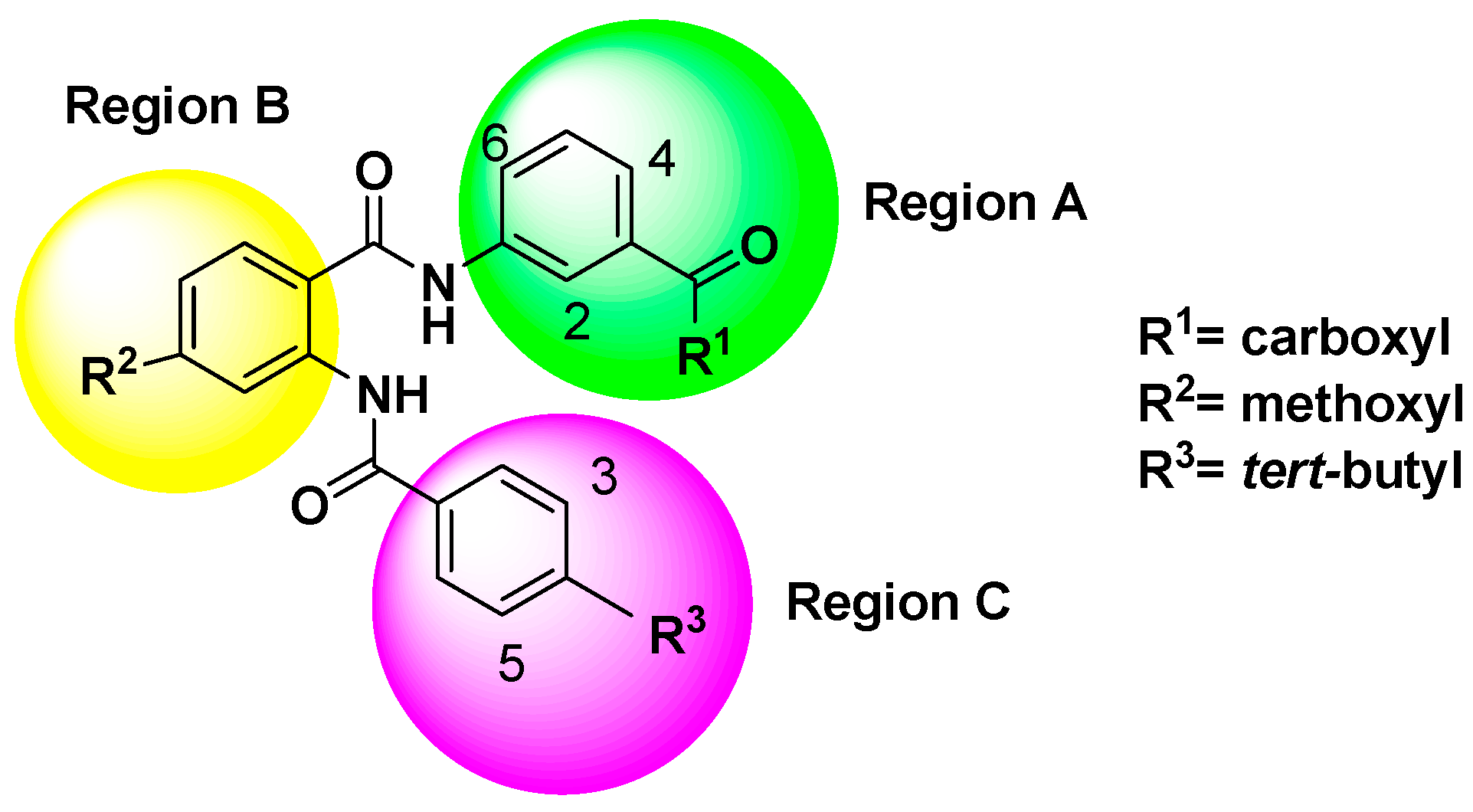
2.3. Design of New Partial FXR Agonists
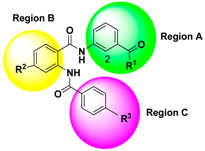
2.3.1. Chemical Structure Design
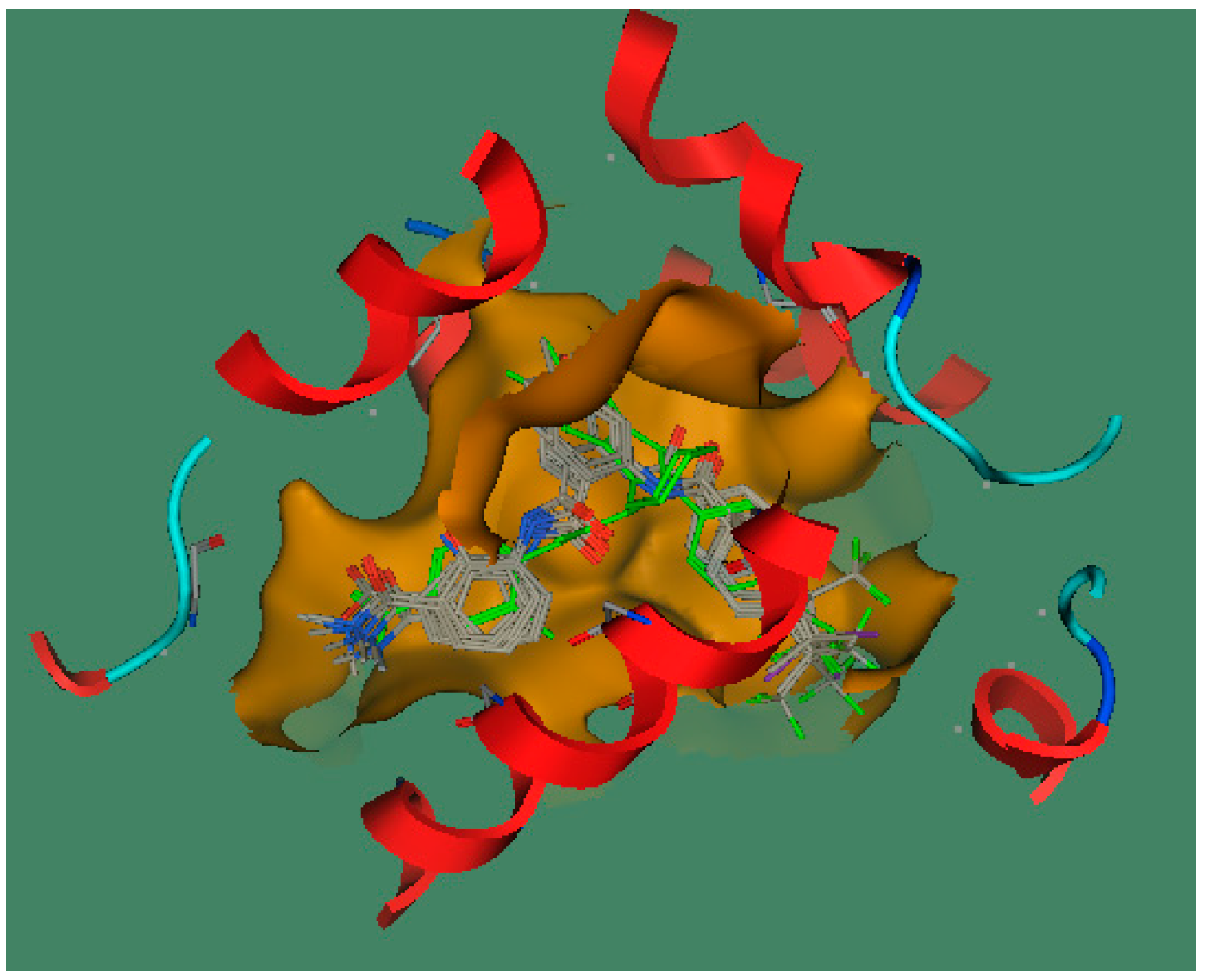
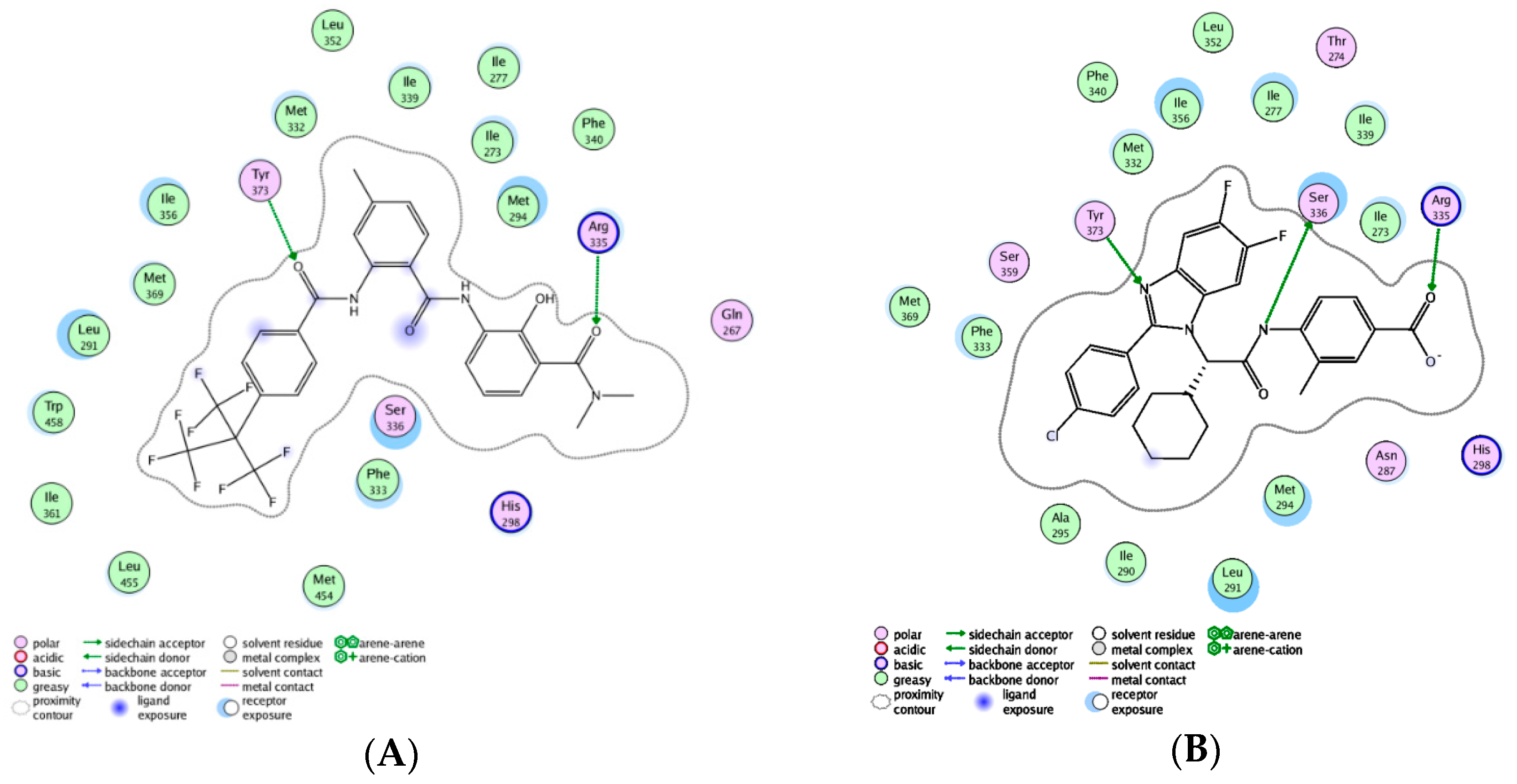
2.3.2. Molecular Docking Study
3. Experimental Section
3.1. Data Set
3.2. 2D-QSAR Studies
3.2.1. Descriptors Calculation
3.2.2. Stepwise Multiple Linear Regression (SW-MLR)
3.3. 3D-QSAR Studies
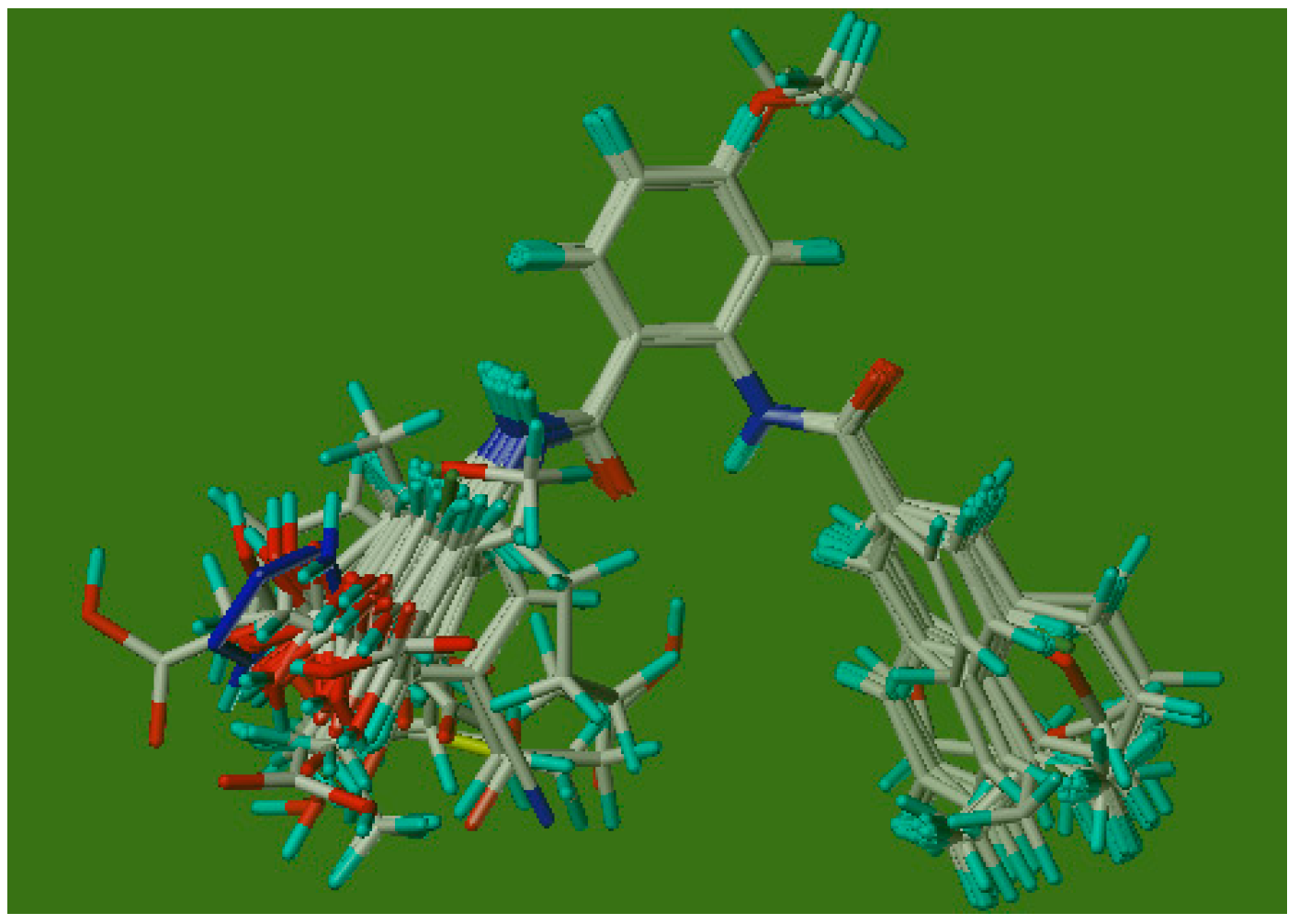
3.3.1. Molecular Alignment
3.3.2. CoMFA Modeling
3.4. Model Validation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Düfer, M.; Hörth, K.; Wagner, R.; Schittenhelm, B.; Prowald, S.; Wagner, T.F.J.; Oberwinkler, J.; Lukowski, R.; Gonzalez, F.J.; Krippeit-Drews, P.; et al. Bile acids acutely stimulate insulin secretion of mouse-cells via farnesoid X receptor activation and KATP channel inhibition. Diabetes 2012, 6, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Nijmeijer, R.M.; Gadaleta, R.M.; van Mil, S.W.C.; van Bodegraven, A.A.; Crusius, J.B.; Dijkstra, G.; Hommes, D.W.; de Jong, D.J.; Stokkers, P.C.F.; Verspaget, H.W.; et al. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PLoS ONE 2011, 8, e23745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollman, D.A.A.; Milona, A.; van Erpecum, K.J.; van Mil, S.W.C. Anti-inflammatory and metabolic actions of FXR: Insights into molecular mechanisms. Biochim. Biophys. Acta 2012, 11, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Akwabi-Ameyaw, A.; Bass, J.Y.; Caldwell, R.D.; Caravella, J.A.; Chen, L. FXR agonist activity of conformationally constrained analogs of GW4064. Bioorg. Med. Chem. Lett. 2009, 19, 4733–4739. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.; Steinhilber, D.; Schubert-Zsilavecz, M. Characterizing ligands for farnesoid X receptor available in vitro test systems for farnesoid X receptor modulator development. Expert Opin. Drug Discov. 2014, 1, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Yadav, M.; Singh, A.K.; Kumar, H.; Dwivedi, S.K. Synthetic FXR agonist GW4064 is a modulator of multiple G protein-coupled receptors. Mol. Endocrinol. 2014, 5, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Flatt, B.; Martin, R.; Wang, T.L.; Mahaney, P.; Murphy, B. Discovery of XL335 (WAY-362450), a highly potent, selective, and orally active agonist of the farnesoid X receptor (FXR). J. Med. Chem. 2009, 4, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.; Gabler, M.; Gomez, R.C.; Flesch, D.; Hanke, T.; Kaiser, A. Anthranilic acid derivatives as novel ligands for farnesoid X receptor(FXR). Bioorg. Med. Chem. 2014, 22, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.; Lamers, C.; Ahmad, K.; Gomez, R.C.; Schneider, G. Extending the structure-activity relationship of anthranilic acid derivatives as farnesoid X receptor modulators: Development of a highly potent partial farnesoid X receptor agonist. J. Med. Chem. 2014, 57, 8035–8055. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V.; Lacrămă, A.M. Introducing spectral structure activity relationship (S-SAR) analysis. Application to ecotoxicology. Int. J. Mol. Sci. 2007, 8, 363–391. [Google Scholar] [CrossRef]
- Putz, M.V.; Putz, A.M.; Lazea, M.; Ienciu, L.; Chiriac, A. Quantum-SAR extension of the spectral-SAR algorithm. Application to polyphenolic anticancer bioactivity. Int. J. Mol. Sci. 2009, 10, 1193–1214. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.D.J.; Vasanthi, A.H.R. QSAR based docking studies of marine algal anticancer compounds as inhibitors of protein kinase B (PKBb). Eur. J. Pharm. Sci. 2015, 76, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Noguera, G.J.; Fabian, L.E.; Lombardo, E.; Finkielsztein, L. QSAR study and conformational analysis of 4-arylthiazolylhydrazones derived from 1-indanones with anti-Trypanosoma cruziactivity. Eur. J. Pharm. Sci. 2015, 78, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Paul, S. Docking and 3D-QSAR studies of acetohydroxy acid synthase inhibitor sulfonylurea derivatives. J. Mol. Model. 2010, 16, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V.; Dudas, N.A.; Isvoran, A. Double variational binding—(SMILES) conformational analysis by docking mechanisms for anti-HIV pyrimidine ligands. Int. J. Mol. Sci. 2015, 16, 19553–19601. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.; Jensen, R.; Heyden, Y.V. QSRR modeling for diverse drugs using different feature selection methods coupled with linear and nonlinear regressions. J. Chromatogr. B 2012, 910, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Jalali-Heravi, M.; Asadollahi-Baboli, M.; Shahbazikhah, P. QSAR study of heparanase inhibitors activity using artificial neural networks and Levenberge-Marquardt algorithm. Eur. J. Med. Chem. 2008, 43, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xiang, Y.; Song, J.; Zhang, Z. A novel two-step QSAR modeling work flow to predict selectivity and activity of HDAC inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xi, L.; Yao, X.; Li, J.Z.; Wu, X. Quantitative structure-activity relationship analysis of a series of human renal organic anion transporter inhibitors. Arch. Pharm. Chem. Life Sci. 2012, 345, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Masand, V.H.; Mahajan, D.T.; Alafeefy, A.M.; Bukhari, S.N.A.; Elsayed, N.N. Optimization of antiproliferative activity of substituted phenyl 4-(2-oxoimidazolidin-1-yl) benzenesulfonates: QSAR and CoMFA analyses. Eur. J. Pharm. Sci. 2015, 77, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Khedr, M.A.; Shehata, T.M.; Mohamed, M.E. Repositioning of 2,4-dichlorophenoxy acetic acid as a potential anti-inflammatory agent: In silico and pharmaceutical formulation study. Eur. J. Pharm. Sci. 2014, 65, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Makhuri, F.R.; Ghasemi, J.B. Computer-aided scaffold hopping to identify a novel series of casein kinase 1 δ (CK1d) inhibitors for amyotrophic lateral sclerosis. Eur. J. Pharm. Sci. 2015, 78, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; Lai, X.M.; Yang, X.M. A QSAR classification study on inhibitory activities of 2-arylbenzoxazoles against cholesteryl ester transfer protein. Med. Chem. Res. 2014, 23, 1878–1886. [Google Scholar] [CrossRef]
- Saghaie, L.; Shahlaei, M.; Fassihi, A.; Madadkar-Sobhani, A.; Gholivand, M.B.; Pourhossein, A. QSAR analysis for some diaryl-substituted pyrazoles as CCR2 Inhibitors by GA-stepwise MLR. Chem. Biol. Drug Des. 2011, 77, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Vrontaki, E.; Melagraki, G.; Mavromoustakos, T.; Afantitis, A. Exploiting ChEMBL database to identify indole analogues as HCV replication inhibitors. Methods 2015, 71, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Fortin, S.; Wei, L.; Moreau, E.; Lacroix, J.; Cote, M.F.; Petitclerc, E.; Kotra, L.P.; Gaudreault, R.C. Substituted phenyl 4-(2-oxoimidazolidin-1-yl)benzenesulfonamides as antimitotics. Antiproliferative, antiangiogenic and antitumoral activity, and quantitative structure–activity relationships. Eur. J. Med. Chem. 2011, 46, 5327–5342. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, M.; Ai, C.; Wang, Y. Insight into the structural determinants of imidazole scaffold-based derivatives as TNF-α release inhibitors by in silico explorations. Int. J. Mol. Sci. 2015, 16, 20118–20138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, B.; Wei, X.; Tan, C.; Chen, Y.; Jiang, Y. A two-step target binding and selectivity support vector machines approach for virtual screening of dopamine receptor subtype-selective ligands. PLoS ONE 2012, 7, e39076. [Google Scholar] [CrossRef] [PubMed]
- Wegner, J.K.; Frohlich, H.; Zell, A. Feature selection for descriptor based classification models. Theory and GA-SEC algorithm. J. Chem. Inf. Comput. Sci. 2004, 44, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Beware of q2. J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Shahlaei, M.; Fassihi, A. QSAR analysis of some 1-(3,3-diphenylpropyl)-piperidinyl amides and ureas as CCR5 inhibitors using genetic algorithm-least square support vector machine. Med. Chem. Res. 2013, 22, 4384–4400. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Chemical Meaning | Coefficient | VIF | Stand Coefficient |
|---|---|---|---|---|
| b_rotN | Number of rotatable single bonds | 0.362 | 2.888 | 0.408 |
| RPC− | Relative negative partial charges | 14.001 | 1.171 | 0.393 |
| opr_leadlike | One if and only if the number of violations of Oprea‘s lead-like test <2 otherwise zero | 0.318 | 1.271 | 0.136 |
| SlogP_VSA2 | The subdivided surface area descriptor, which is based on sum of the approximate accessible van der Waal’s surface area | −0.049 | 3.728 | −0.567 |
| ASA | Water accessible surface area calculated using a radius of 1.4 A for the water molecule | 0.016 | 1.539 | 0.848 |
| Constant | - | −9.717 | – | – |
| Descriptor | b_rotN | RPC− | opr_leadlike | SlogP_VSA2 | ASA |
|---|---|---|---|---|---|
| b_rotN | 1.000 | 0.359 | 0.137 | 0.715 | −0.032 |
| RPC− | 0.359 | 1.000 | −0.010 | 0.215 | −0.289 |
| opr_leadlike | 0.137 | −0.010 | 1.000 | 0.298 | −0.461 |
| SlogP_VSA2 | 0.715 | 0.215 | 0.298 | 1.000 | −0.366 |
| ASA | −0.032 | −0.289 | −0.461 | −0.366 | 1.000 |
 | |||||||
|---|---|---|---|---|---|---|---|
| NO. | R1 | R2 | R3 | EC50 (µM) | pEC50 | 2D-Pred | 3D-Pred |
| SW-MLR | CoMFA | ||||||
| 1 | 3-carboxyphenyl | H | 4-tert-butylphenyl | 0.28 | 6.553 | 6.784 | 6.704 |
| 2 * | 3-carboxyphenyl | H | 4-(trifluoromethyl)phenyl | 6.9 | 5.161 | 5.653 | 5.171 |
| 3 | 3-carboxyphenyl | H | 4-bromophenyl | 3.7 | 5.432 | 5.451 | 5.485 |
| 4 | 3-carboxyphenyl | H | benzo[d][1,3]dioxol-5-yl | 10 | 5.000 | 5.336 | 5.133 |
| 5 * | 3-carboxyphenyl | H | 2,3-dihydrobenzo[b][1,4]dioxin-6-yl | 4.9 | 5.310 | 5.714 | 5.133 |
| 6 | 3-carboxyphenyl | H | 3-fluoro-4-(trifluoromethyl)phenyl | 5 | 5.301 | 5.232 | 5.358 |
| 7 * | 3-carboxyphenyl | H | styryl | 5.2 | 5.284 | 4.850 | 5.639 |
| 8 | 3-acetylphenyl | H | 4-tert-butylphenyl | 0.48 | 6.319 | 6.592 | 6.775 |
| 9 | 3-cyanophenyl | H | 4-tert-butylphenyl | 0.23 | 6.638 | 6.619 | 6.506 |
| 10 | 3-methoxyphenyl | H | 4-tert-butylphenyl | 0.38 | 6.420 | 6.661 | 6.327 |
| 11 | 3-(methylthio)phenyl | H | 4-tert-butylphenyl | 0.2 | 6.699 | 6.616 | 6.655 |
| 12 | 3-(1H-tetrazol-5-yl)phenyl | H | 4-tert-butylphenyl | 2.9 | 5.538 | 5.854 | 5.695 |
| 13 | 3-carbamoylphenyl | H | 4-tert-butylphenyl | 0.074 | 7.131 | 7.203 | 7.099 |
| 14 * | 3,4-bimethoxyphenyl | H | 4-tert-butylphenyl | 0.071 | 7.149 | 7.771 | 6.606 |
| 15 | 3-(carboxymethyl)phenyl | H | 4-tert-butylphenyl | 0.42 | 6.377 | 6.493 | 6.414 |
| 16 | 3-(2-carboxyethyl)phenyl | H | 4-tert-butylphenyl | 0.064 | 7.194 | 7.143 | 6.999 |
| 17 | 2-methyl-3-carboxylphenyl | H | 4-tert-butylphenyl | 0.042 | 7.377 | 7.115 | 7.266 |
| 18 | 2-methoxy-5-carboxyphenyl | H | 4-tert-butylphenyl | 4.7 | 5.328 | 5.307 | 5.444 |
| 19 | 2-fluoro-5-carboxyphenyl | H | 4-tert-butylphenyl | 0.48 | 6.319 | 6.148 | 6.361 |
| 20* | 2-chloro-5-carboxyphenyl | H | 4-tert-butylphenyl | 1.1 | 5.959 | 6.569 | 5.759 |
| 21 | 3-carboxy-4-methylphenyl | H | 4-tert-butylphenyl | 0.045 | 7.347 | 7.209 | 6.934 |
| 22 * | 3-carboxy-4-methoxylphenyl | H | 4-tert-butylphenyl | 0.047 | 7.328 | 7.650 | 7.563 |
| 23 | 3-carboxy-4-chlorophenyl | H | 4-tert-butylphenyl | 0.28 | 6.553 | 6.845 | 6.931 |
| 24 | 3-carboxy-4-bromophenyl | H | 4-tert-butylphenyl | 0.15 | 6.824 | 6.937 | 6.889 |
| 25 | 3-carboxyphenyl | chloro | 4-tert-butylphenyl | 0.047 | 7.328 | 6.862 | 6.868 |
| 26 * | 4-carboxymethylphenyl | H | naphthalen-2-yl | 3.1 | 5.509 | 5.492 | 5.082 |
| 27 | 3-carboxy-4-methylphenyl | methyl | 4-tert-butylphenyl | 0.043 | 7.367 | 7.048 | 7.510 |
| 28 | 3-carboxyphenyl | methyl | 4-tert-butylphenyl | 0.061 | 7.215 | 7.334 | 6.942 |
| 29 | 3-carboxyphenyl | bromo | 4-tert-butylphenyl | 0.048 | 7.319 | 7.160 | 7.241 |
| 30 | 3-carboxyphenyl | methoxy | 4-tert-butylphenyl | 0.008 | 8.097 | 7.888 | 8.175 |
| 31 | 3-carboxy-4-methylphenyl | chloro | 4-tert-butylphenyl | 0.11 | 6.959 | 7.178 | 7.027 |
| 32 * | 3-carboxy-4-methylphenyl | methoxy | 4-tert-butylphenyl | 0.087 | 7.060 | 7.861 | 6.858 |
| 33 | 3-carboxypropyl | H | 4-ehylphenyl | 5.8 | 5.237 | 5.058 | 5.253 |
| 34 | 3-carboxypropyl | H | 4-tert-butylphenyl | 2.5 | 5.602 | 5.748 | 5.885 |
| 35 | 3-carboxypropyl | H | naphthalen-2-yl | 8.6 | 5.066 | 4.598 | 4.817 |
| 36 | 4-carboxybutyl | H | naphthalen-2-yl | 8.3 | 5.081 | 5.086 | 4.795 |
| 37 * | 3-methoxy-3-oxopropyl | H | naphthalen-2-yl | 7.1 | 5.149 | 5.377 | 4.857 |
| 38 | 5-carboxypentyl | H | naphthalen-2-yl | 4.4 | 5.357 | 5.657 | 5.416 |
| 39 | 4-carboxyphenyl | H | naphthalen-2-yl | 1.0 | 6.000 | 5.882 | 5.902 |
| 40 | 3-carboxyphenyl | H | naphthalen-2-yl | 1.5 | 5.824 | 5.750 | 5.990 |
| 41 * | 4-carboxybenzyl | H | naphthalen-2-yl | 1.3 | 5.886 | 6.722 | 5.405 |
| Model | Training Set | Test Set | |||||
|---|---|---|---|---|---|---|---|
| R2train | RMSEtrain | F | Q2LOO | RMSELOO | R2test | RMSEtest | |
| MLR | 0.935 | 0.219 | 72.353 | 0.899 | 0.299 | 0.902 | 0.534 |
| CoMFA-1 | 0.944 | 0.203 | 109.711 | 0.802 | 0.383 | 0.892 | 0.330 |
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| R2train | 0.150 | 0.145 | 0.188 | 0.08 | 0.123 | 0.179 | 0.144 | 0.186 | 0.141 | 0.131 |
| Q2LOO | 0.013 | 0.058 | 0.016 | 0.039 | 0.014 | 0.012 | 0.105 | 0.000 | 0.005 | 0.014 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Name | R1 | Substituents at Position 2 | R2 | R3 | Predicted pEC50 Values | Docking Scores | |
| SW-MLR | CoMFA | ||||||
| T30 | OH | H | OCH3 | C(CH3)3 | 7.888 | 8.175 | −10.176 |
| N1 | N(CH3)2 | H | CH3 | C(CF3)3 | 9.032 | 8.350 | −14.053 |
| N2 | N(CH3)2 | H | CH3 | C(CH3)3 | 8.274 | 8.323 | −11.081 |
| N3 | N(CH3)2 | H | OCH3 | C(CH3)3 | 8.626 | 8.322 | −10.716 |
| N4 | N(CH3)2 | H | CH3 | CI3 | 8.744 | 8.304 | −12.038 |
| N5 | N(CH3)2 | OH | CH3 | C(CH3)3 | 8.260 | 8.360 | −11.602 |
| N6 | N(CH3)2 | OH | CH3 | CI3 | 8.828 | 8.357 | −12.073 |
| N7 | N(CH3)2 | CH3 | CH3 | C(CF3)3 | 9.123 | 8.374 | −14.193 |
| N8 | N(CH3)2 | NH2 | CH3 | C(CF3)3 | 8.816 | 8.378 | −14.335 |
| N9 | N(CH3)2 | OH | CH3 | C(CF3)3 | 9.024 | 8.388 | −14.347 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Yang, X.; Lai, X.; Kang, J.; Gan, H.; Gao, Y. Structural Investigation for Optimization of Anthranilic Acid Derivatives as Partial FXR Agonists by in Silico Approaches. Int. J. Mol. Sci. 2016, 17, 536. https://doi.org/10.3390/ijms17040536
Chen M, Yang X, Lai X, Kang J, Gan H, Gao Y. Structural Investigation for Optimization of Anthranilic Acid Derivatives as Partial FXR Agonists by in Silico Approaches. International Journal of Molecular Sciences. 2016; 17(4):536. https://doi.org/10.3390/ijms17040536
Chicago/Turabian StyleChen, Meimei, Xuemei Yang, Xinmei Lai, Jie Kang, Huijuan Gan, and Yuxing Gao. 2016. "Structural Investigation for Optimization of Anthranilic Acid Derivatives as Partial FXR Agonists by in Silico Approaches" International Journal of Molecular Sciences 17, no. 4: 536. https://doi.org/10.3390/ijms17040536