
Insights into the Antimicrobial Mechanism of Action of Human RNase6: Structural Determinants for Bacterial Cell Agglutination and Membrane Permeation

Abstract
:
1. Introduction
2. Results
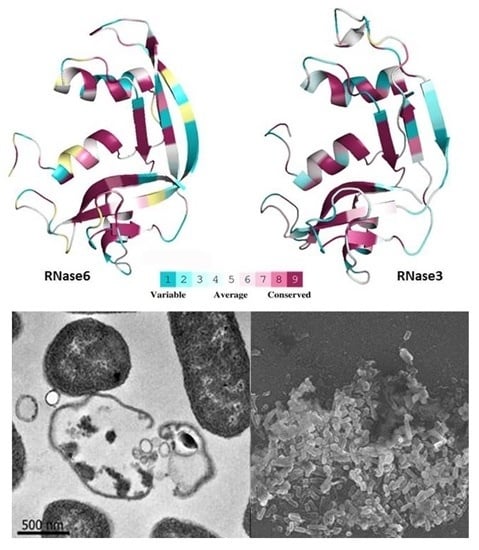
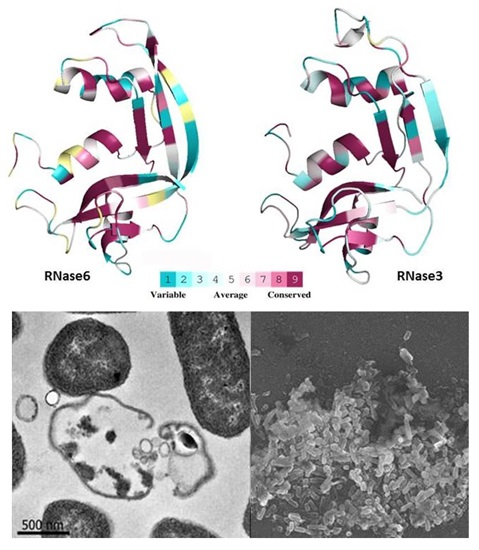
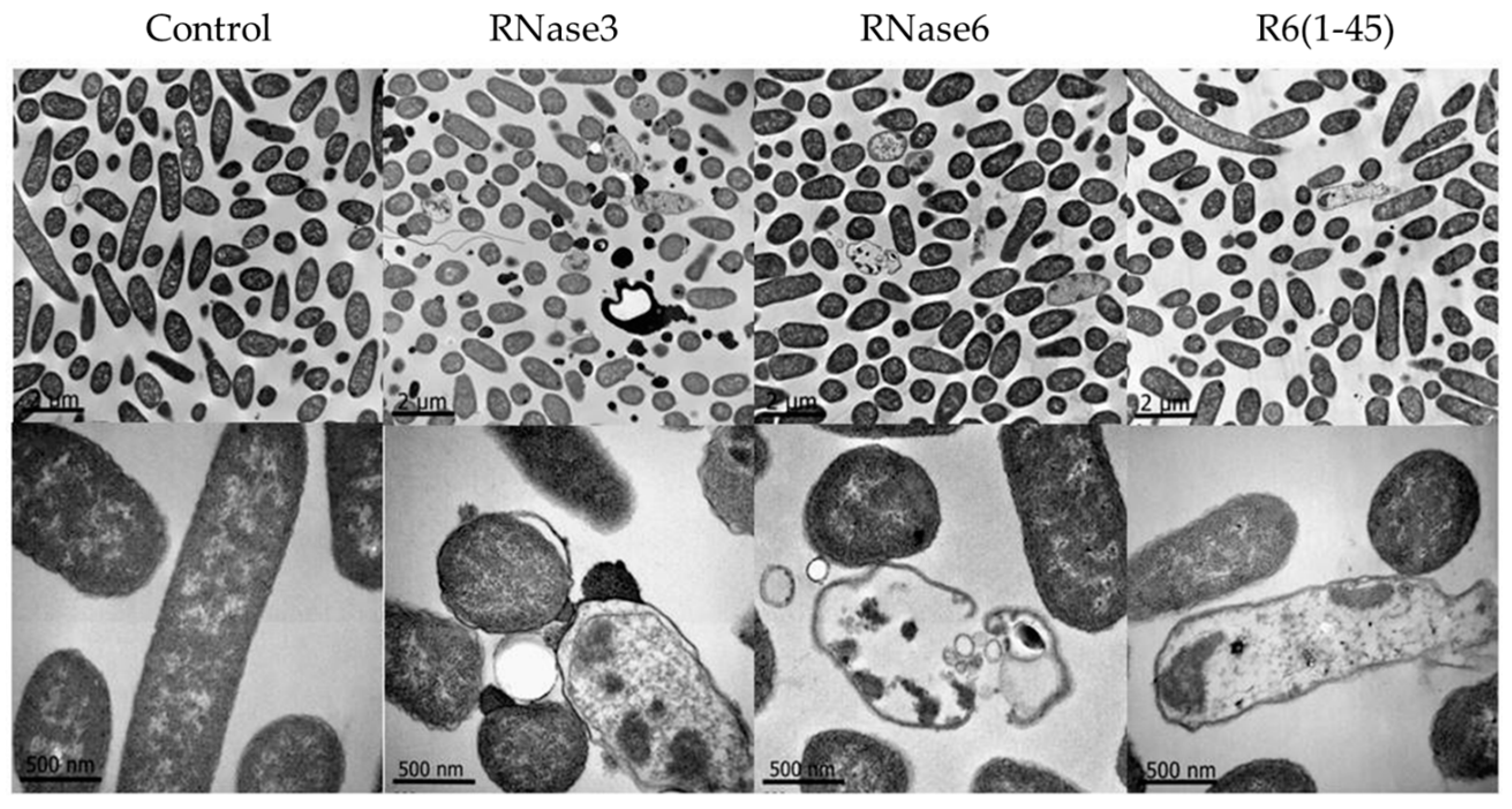
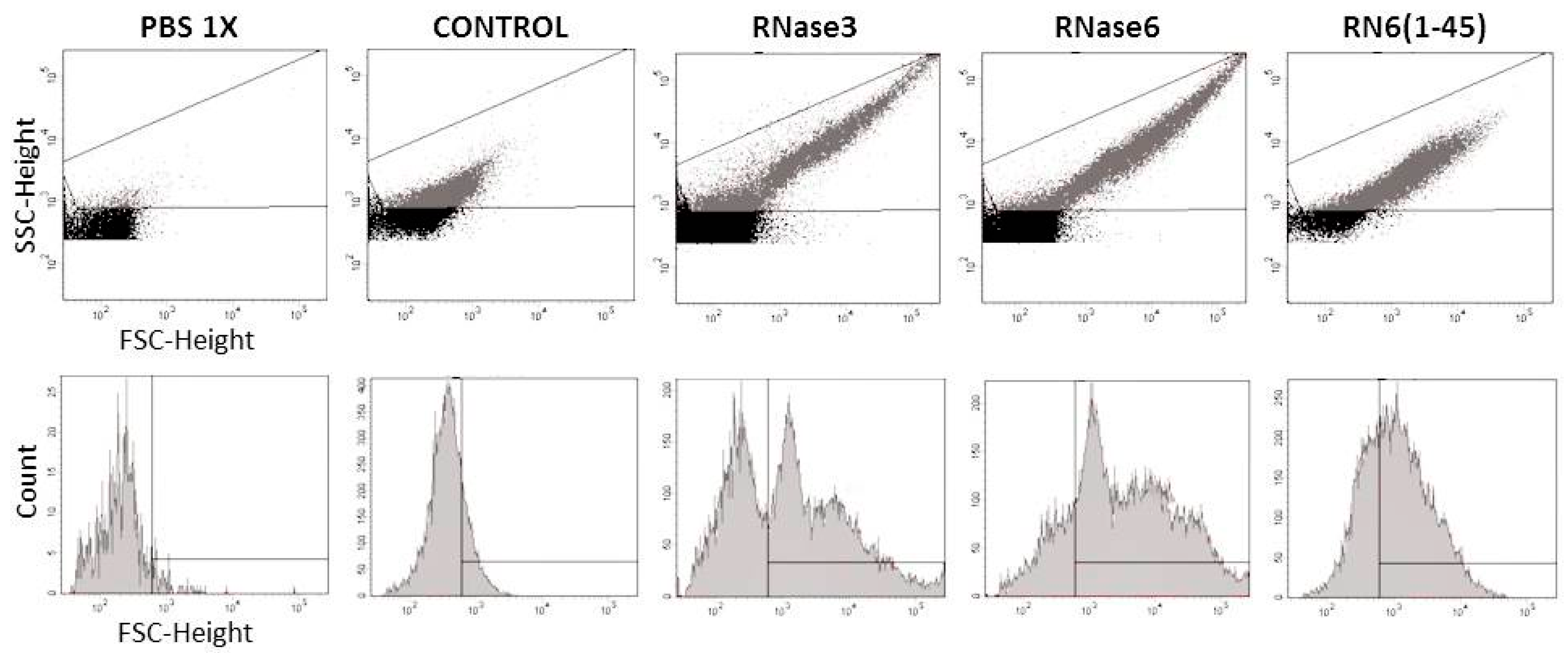
2.1. Membrane and Cell Wall Interaction
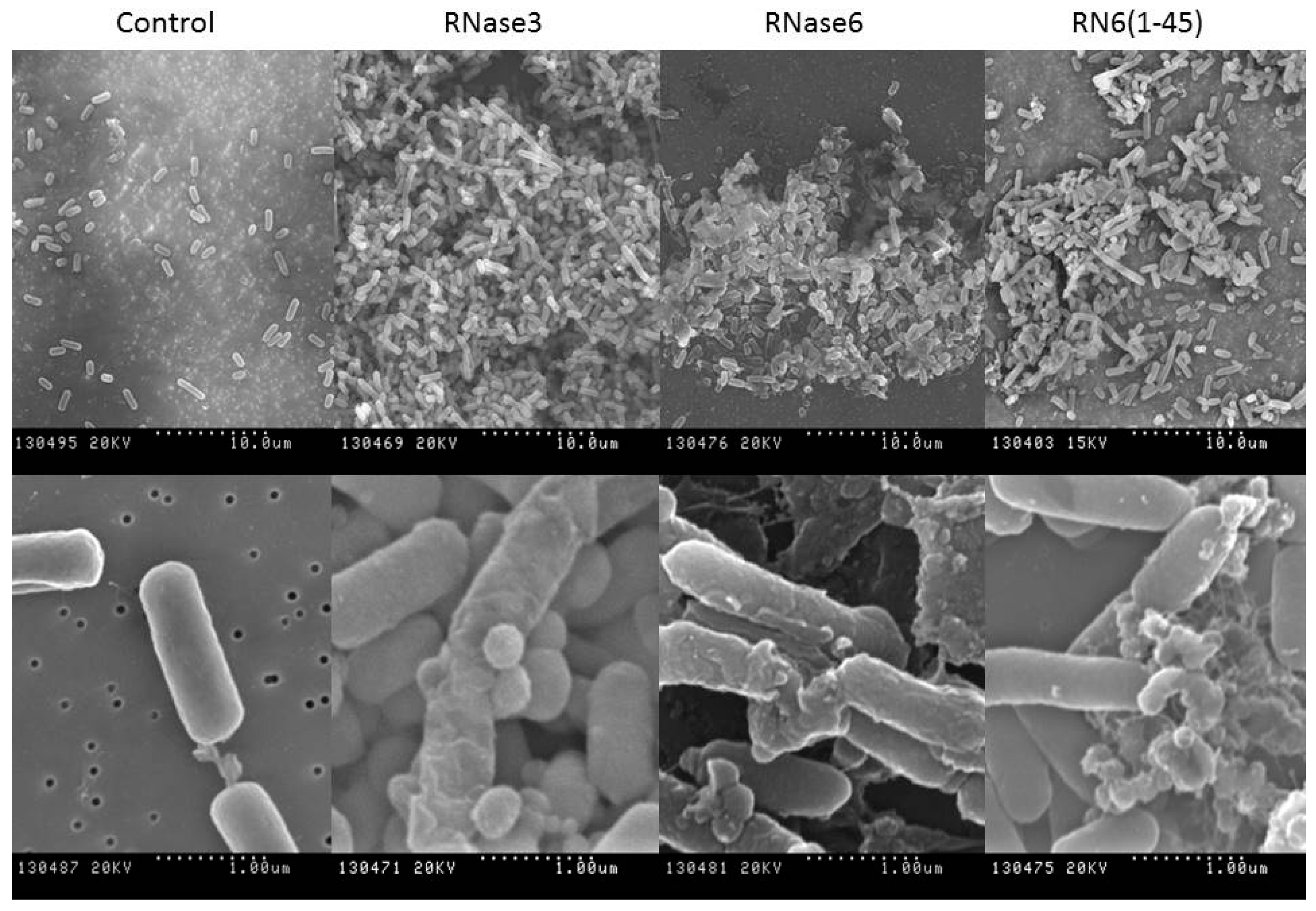
2.2. Bactericidal Activity
2.3. Mutant Design and Characterization
3. Discussion
4. Materials and Methods
4.1. Materials and Strains
4.2. Protein Expression and Purification
4.3. Minimal Bactericidal Concentration (MBC)
4.4. Bacterial Viability Assays
4.5. Bacterial Cell Membrane Depolarization Assay
4.6. Minimal Agglutination Activity (MAC)
4.7. Fluorescence Activated Cell-Sorting (FACS)
4.8. Scanning Electron Microscopy (SEM)
4.9. Transmission Electron Microscopy
4.10. Hemolytic Activity
4.11. Liposome Preparation
4.12. Intrinsic Tryptophan Fluorescence Analysis
4.13. Liposome Leakage
4.14. Liposome Aggregation
4.15. LPS Binding Fluorimetric Assay
4.16. Aggrescan3D Analysis
4.17. Minimal Inhibitory Concentration (MIC)
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ANTS | 8-aminonaphthalene-1,3,6-trisulfonic acid |
| BC | BODIPY® cadaverine |
| CFU | colony forming units |
| DLS | dynamic light scattering |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| DOPG | 1,2-dioleoyl-sn-glycero-3-phosphoglycerol |
| DPX | p-xylenebispyridinium bromide |
| ED50 | 50% effective dose |
| FACS | fluorescent activated cell-sorting |
| HC | hemolytic activity |
| LB | Luria-Bertani media |
| LPS | lipopolysaccharide |
| LUV | large unilamellar vesicle |
| MAC | minimal agglutination concentration |
| MIC | minimal inhibitory concentration |
| PBS | phosphate buffer saline |
| RBC | red blood cell |
| RNase | Ribonuclease |
| SDS | sodium dodecyl sulfate |
| SEM | scanning electron microscopy |
| TB | terrific broth media |
| TEM | transmission electron microscopy |
References
- Beintema, J.J.; Kleineidam, R.G. The ribonuclease a superfamily: General discussion. Cell. Mol. Life Sci. 1998, 54, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Goo, S.M.; Cho, S. The expansion and functional diversification of the mammalian ribonuclease a superfamily epitomizes the efficiency of multigene families at generating biological novelty. Genome Biol. Evol. 2013, 5, 2124–2140. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Pulido, D.; Valle, J.; Nogues, M.V.; Andreu, D.; Boix, E. Ribonucleases as a host-defence family: Evidence of evolutionarily conserved antimicrobial activity at the N-terminus. Biochem. J. 2013, 456, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Beintema, J.J.; Zhang, J. The ribonuclease a superfamily of mammals and birds: Identifying new members and tracing evolutionary histories. Genomics 2005, 85, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, E.; Merlino, A.; Turano, M.; Russo Krauss, I.; Coscia, F.; Zanfardino, A.; Varcamonti, M.; Furia, A.; Giancola, C.; Mazzarella, L.; et al. A new RNase sheds light on the RNase/angiogenin subfamily from zebrafish. Biochem. J. 2011, 433, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S. The eight human “Canonical” Ribonucleases: Molecular diversity, catalytic properties, and special biological actions of the enzyme proteins. FEBS Lett. 2010, 584, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Boix, E.; Nogues, M.V. Mammalian antimicrobial proteins and peptides: Overview on the RNase a superfamily members involved in innate host defence. Mol. Biosyst. 2007, 3, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, E.; D’Alessio, G. The success of the RNase scaffold in the advance of biosciences and in evolution. Gene 2007, 406, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.F. RNase a ribonucleases and host defense: An evolving story. J. Leukoc. Biol. 2008, 83, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Haigh, B.J.; Griffin, F.J.; Wheeler, T.T. The mammalian secreted RNases: Mechanisms of action in host defence. Innate Immun. 2013, 19, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Batra, J.K. Antimicrobial activity of human eosinophil granule proteins: Involvement in host defence against pathogens. Crit. Rev. Microbiol. 2012, 38, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.R.; Ackerman, S.J. Eosinophil granule proteins: Form and function. J. Biol. Chem. 2014, 289, 17406–17415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Rosenberg, H.F.; Nei, M. Positive darwinian selection after gene duplication in primate ribonuclease genes. Proc. Natl. Acad. Sci. USA 1998, 95, 3708–3713. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.F.; Dyer, K.D.; Tiffany, H.L.; Gonzalez, M. Rapid evolution of a unique family of primate ribonuclease genes. Nat. Genet. 1995, 10, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Domachowske, J.B.; Dyer, K.D.; Bonville, C.A.; Rosenberg, H.F. Recombinant human eosinophil-derived neurotoxin/RNase 2 functions as an effective antiviral agent against respiratory syncytial virus. J. Infect. Dis. 1998, 177, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.F. Eosinophil-derived neurotoxin/RNase2: Connecting the past, the present and the future. Curr. Pharm. Biotechnol. 2008, 9, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.F. Eosinophil-derived neurotoxin (EDN/RNase2) and the mouse eosinophil-associated RNases (mEars): Expanding roles in promoting host defense. Int. J. Mol. Sci. 2015, 16, 15442–15455. [Google Scholar] [CrossRef] [PubMed]
- Carreras, E.; Boix, E.; Rosenberg, H.F.; Cuchillo, C.M.; Nogues, M.V. Both aromatic and cationic residues contribute to the membrane-lytic and bactericidal activity of eosinophil cationic protein. Biochemistry 2003, 42, 6636–6644. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Pulido, D.; Nogues, M.V.; Boix, E. Exploring new biological functions of amyloids: Bacteria cell agglutination mediated by host protein aggregation. PLoS Pathog. 2012, 8, e1003005. [Google Scholar] [CrossRef] [PubMed]
- Lien, P.C.; Kuo, P.H.; Chen, C.J.; Chang, H.H.; Fang, S.L.; Wu, W.S.; Lai, Y.K.; Pai, T.W.; Chang, M.D. In silico prediction and in vitro characterization of multifunctional human RNase3. Biomed Res. Int. 2013, 2013, 170398. [Google Scholar] [CrossRef] [PubMed]
- Boix, E.; Salazar, V.A.; Torrent, M.; Pulido, D.; Nogues, M.V.; Moussaoui, M. Structural determinants of the eosinophil cationic protein antimicrobial activity. Biol. Chem. 2012, 393, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Batra, J.K. Role of unique basic residues in cytotoxic, antibacterial and antiparasitic activities of human eosinophil cationic protein. Biol. Chem. 2011, 392, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.F. Recombinant human eosinophil cationic protein. Ribonuclease activity is not essential for cytotoxicity. J. Biol. Chem. 1995, 270, 7876–7881. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.D.; Schwaderer, A.L.; Dirosario, J.D.; McHugh, K.M.; McGillivary, G.; Justice, S.S.; Carpenter, A.R.; Baker, P.B.; Harder, J.; Hains, D.S. Ribonuclease 7 is a potent antimicrobial peptide within the human urinary tract. Kidney Int. 2011, 80, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Schroder, J.M. RNase7, a novel innate immune defense antimicrobial protein of healthy human skin. J. Biol. Chem. 2002, 277, 46779–46784. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Song, P.I.; Seo, C.H.; Cheong, H.; Park, Y. Colonization and infection of the skin by S. Aureus: Immune system evasion and the response to cationic antimicrobial peptides. Int. J. Mol. Sci. 2014, 15, 8753–8772. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Schwaderer, A.L.; Kline, J.; Spencer, J.D.; Kline, D.; Hains, D.S. Contribution of structural domains to the activity of ribonuclease 7 against uropathogenic bacteria. Antimicrob. Agents Chemother. 2013, 57, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.D.; Schwaderer, A.L.; Wang, H.; Bartz, J.; Kline, J.; Eichler, T.; DeSouza, K.R.; Sims-Lucas, S.; Baker, P.; Hains, D.S. Ribonuclease 7, an antimicrobial peptide upregulated during infection, contributes to microbial defense of the human urinary tract. Kidney Int. 2013, 83, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.F.; Dyer, K.D. Molecular cloning and characterization of a novel human ribonuclease (RNase k6): Increasing diversity in the enlarging ribonuclease gene family. Nucleic Acids Res. 1996, 24, 3507–3513. [Google Scholar] [CrossRef] [PubMed]
- Becknell, B.; Eichler, T.E.; Beceiro, S.; Li, B.; Easterling, R.S.; Carpenter, A.R.; James, C.L.; McHugh, K.M.; Hains, D.S.; Partida-Sanchez, S.; et al. Ribonucleases 6 and 7 have antimicrobial function in the human and murine urinary tract. Kidney Int. 2014, 87, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Boix, E.; Torrent, M.; Sanchez, D.; Nogues, M.V. The antipathogen activities of eosinophil cationic protein. Curr. Pharm. Biotechnol. 2008, 9, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Amatngalim, G.D.; van Wijck, Y.; de Mooij-Eijk, Y.; Verhoosel, R.M.; Harder, J.; Lekkerkerker, A.N.; Janssen, R.A.; Hiemstra, P.S. Basal cells contribute to innate immunity of the airway epithelium through production of the antimicrobial protein RNase 7. J. Immunol. 2015, 194, 3340–3350. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new endscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Prats-Ejarque, G.; Arranz-Trullen, J.; Blanco, J.A.; Pulido, D.; Nogues, M.V.; Moussaoui, M.; Boix, E. The first crystal structure of human rnase6 reveals a novel substrate binding and cleavage site arrangement. Biochem. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Boix, E.; Pulido, D.; Moussaoui, M.; Nogues, M.V.; Russi, S. The sulfate-binding site structure of the human eosinophil cationic protein as revealed by a new crystal form. J. Struct. Biol. 2012, 179, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. Consurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Navarro, S.; Moussaoui, M.; Nogues, M.V.; Boix, E. Eosinophil cationic protein high-affinity binding to bacteria-wall lipopolysaccharides and peptidoglycans. Biochemistry 2008, 47, 3544–3555. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; de la Torre, B.G.; Nogues, V.M.; Andreu, D.; Boix, E. Bactericidal and membrane disruption activities of the eosinophil cationic protein are largely retained in an N-terminal fragment. Biochem. J. 2009, 421, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Sanchez, D.; Buzon, V.; Nogues, M.V.; Cladera, J.; Boix, E. Comparison of the membrane interaction mechanism of two antimicrobial RNases: RNase3/ECP and RNase7. Biochim. Biophys. Acta 2009, 1788, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Odorizzi, F.; Nogues, M.V.; Boix, E. Eosinophil cationic protein aggregation: Identification of an N-terminus amyloid prone region. Biomacromolecules 2010, 11, 1983–1990. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Badia, M.; Moussaoui, M.; Sanchez, D.; Nogues, M.V.; Boix, E. Comparison of human RNase3 and RNase7 bactericidal action at the Gram-negative and Gram-positive bacterial cell wall. FEBS J. 2010, 277, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Pulido, D.; Moussaoui, M.; Andreu, D.; Nogues, M.V.; Torrent, M.; Boix, E. Antimicrobial action and cell agglutination by the eosinophil cationic protein are modulated by the cell wall lipopolysaccharide structure. Antimicrob. Agents Chemother. 2012, 56, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Cuyas, E.; Carreras, E.; Navarro, S.; Lopez, O.; de la Maza, A.; Nogues, M.V.; Reshetnyak, Y.K.; Boix, E. Topography studies on the membrane interaction mechanism of the eosinophil cationic protein. Biochemistry 2007, 46, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Pulido, D.; de la Torre, B.G.; Garcia-Mayoral, M.F.; Nogues, M.V.; Bruix, M.; Andreu, D.; Boix, E. Refining the eosinophil cationic protein antibacterial pharmacophore by rational structure minimization. J. Med. Chem. 2011, 54, 5237–5244. [Google Scholar] [CrossRef] [PubMed]
- Pulido, D.; Moussaoui, M.; Nogues, M.V.; Torrent, M.; Boix, E. Towards the rational design of antimicrobial proteins: Single point mutations can switch on bactericidal and agglutinating activities on the RNase a superfamily lineage. FEBS J. 2013, 280, 5841–5852. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo, C.M.; Nogues, M.V.; Raines, R.T. Bovine pancreatic ribonuclease: Fifty years of the first enzymatic reaction mechanism. Biochemistry 2011, 50, 7835–7841. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; di Tommaso, P.; Pulido, D.; Nogues, M.V.; Notredame, C.; Boix, E.; Andreu, D. AMPA: An automated web server for prediction of protein antimicrobial regions. Bioinformatics 2011, 28, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Nogues, M.V.; Boix, E. Eosinophil cationic protein (ECP) can bind heparin and other glycosaminoglycans through its RNase active site. J. Mol. Recognit. 2010, 24, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.; Moussaoui, M.; Carreras, E.; Torrent, M.; Nogues, V.; Boix, E. Mapping the eosinophil cationic protein antimicrobial activity by chemical and enzymatic cleavage. Biochimie 2011, 93, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Gorr, S.U.; Sotsky, J.B.; Shelar, A.P.; Demuth, D.R. Design of bacteria-agglutinating peptides derived from parotid secretory protein, a member of the bactericidal/permeability increasing-like protein family. Peptides 2008, 29, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Beintema, J.J. Introduction: The ribonuclease a superfamily. Cell. Mol. Life Sci. 1998, 54, 763–765. [Google Scholar] [CrossRef] [PubMed]
- Deming, M.S.; Dyer, K.D.; Bankier, A.T.; Piper, M.B.; Dear, P.H.; Rosenberg, H.F. Ribonuclease k6: Chromosomal mapping and divergent rates of evolution within the RNase a gene superfamily. Genome Res. 1998, 8, 599–607. [Google Scholar] [PubMed]
- Dyer, K.D.; Rosenberg, H.F. The RNase A superfamily: Generation of diversity and innate host defense. Mol. Divers. 2006, 10, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Boix, E.; Nikolovski, Z.; Moiseyev, G.P.; Rosenberg, H.F.; Cuchillo, C.M.; Nogues, M.V. Kinetic and product distribution analysis of human eosinophil cationic protein indicates a subsite arrangement that favors exonuclease-type activity. J. Biol. Chem. 1999, 274, 15605–15614. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.T.; Lee, J.Y.; Kim, H.J.; Eu, Y.J.; Shin, S.Y.; Hahm, K.S.; Kim, J.I. Contribution of a central proline in model amphipathic alpha-helical peptides to self-association, interaction with phospholipids, and antimicrobial mode of action. FEBS J. 2006, 273, 4040–4054. [Google Scholar] [CrossRef] [PubMed]
- Pulido, D.; Torrent, M.; Andreu, D.; Nogues, M.V.; Boix, E. Two human host defense ribonucleases against mycobacteria, the eosinophil cationic protein (RNase3) and RNase7. Antimicrob. Agents Chemother. 2013, 57, 3797–3805. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein/Peptide | λmax for Fluorescence Emission (nm) | ||||
|---|---|---|---|---|---|
| Buffer | DOPC a | DOPG a | DOPC:DOPG(3:2) a | LPS a | |
| RNase3 | 343 | - | 3 | 3 | 4 |
| RNase6 | 345 | - | 2 | 6 | 4 |
| RN6(1–45) | 343 | - | 8 | 9 | 3 |
| Protein/Peptide | LPS Binding | |
|---|---|---|
| ED50 (µM) a | Max (%) * | |
| RNase3 | 2.54 ± 0.14 | 100 |
| RNase6 | 2.83 ± 0.22 | 76 |
| RN6(1–45) | 5.31 ± 1.41 | 27 |
| Protein/Peptide | Liposome Leakage (µM) | |
|---|---|---|
| ED50 (µM) a | Max (%) * | |
| RNase3 | 0.7 ± 0.1 | 100 |
| RNase6 | 1.5 ± 0.5 | 100 |
| RN6(1–45) | 4.0 ± 0.5 | 100 |
| Protein/Peptide | MBC100 (μm) a | ||||||
|---|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | A. baumanii | S. aureus | M. luteus | E. faecium | HC50 (μm) * | |
| RNase3 | 0.35 ± 0.02 | 0.20 ± 0.01 | 0.40 ± 0.03 | 0.40 ± 0.03 | 0.65 ± 0.08 | 0.90 ± 0.14 | >20 |
| RNase6 | 0.90 ± 0.14 | 0.90 ± 0.14 | 0.65 ± 0.08 | 1.87 ± 0.56 | 1.35 ± 0.23 | 1.35 ± 0.23 | >20 |
| RN6(1–45) | 1.35 ± 0.23 | 0.40 ± 0.03 | 0.65 ± 0.08 | 1.87 ± 0.56 | 1.35 ± 0.23 | 1.35 ± 0.23 | >20 |
| Protein/Peptide | Depolarization (µM) | |||
|---|---|---|---|---|
| E. coli | S. aureus | |||
| ED50 | Depolmax * | ED50 | Depolmax * | |
| RNase3 | 0.5 ± 0.1 | 100.5 ± 3.8 | 0.7 ± 0.2 | 100.5 ± 6.7 |
| RNase6 | 0.6 ± 0.1 | 64.1 ± 4.2 | 0.9 ± 0.1 | 71.7 ± 1.5 |
| RN6(1–45) | 1.1 ± 0.3 | 69.7 ± 5.3 | 1.2 ± 0.2 | 76.2 ± 5.6 |
| Protein/Peptide | Bacterial Viability Assay | |||
|---|---|---|---|---|
| E. coli | S. aureus | |||
| t50 (min) * | Viability (%) * | t50 (min) * | Viability (%) * | |
| RNase3 | 10.6 ± 0.1 | 4.2 ± 0.3 | 7.1 ± 0.1 | 9.3 ± 0.2 |
| RNase6 | 15.4 ± 0.1 | 8.8 ± 0.1 | 18.0 ± 0.4 | 12.9 ± 0.8 |
| RN6(1–45) | 13.1 ± 0.1 | 5.2 ± 0.1 | 5.5 ± 0.1 | 5.8 ± 0.2 |
| Protein/Peptide | MAC (µM) a | Agglutination Activity (%) * |
|---|---|---|
| RNase3 | 0.20 ± 0.05 | 60.35 ± 0.50 |
| RNase6 | 0.20 ± 0.05 | 80.64 ± 0.50 |
| RN6(1–45) | 5 ± 0.50 | 66.22 ± 0.50 |
| Protein/Peptide | MBC100(µM) a | MAC (µM) | LPS Binding Assay | |||
|---|---|---|---|---|---|---|
| E. coli | S. aureus | E. coli | S. aureus | ED50 (µM) | Max (%) * | |
| RNase3 | 0.35 ± 0.01 | 0.40 ± 0.10 | 0.22 ± 0.01 | >5 | 2.54 ± 0.16 | 99.89 ± 4.20 |
| RNase6 | 0.90 ± 0.14 | 1.87 ± 0.56 | 0.22 ± 0.01 | >5 | 2.63 ± 0.23 | 75.89 ± 4.41 |
| RNase6-W1A | 1.87 ± 0.56 | 3.75 ± 0.78 | >5 | >5 | 3.37 ± 0.34 | 38.55 ± 2.32 |
| RNase6-I13A | 3.75 ± 0.78 | >5 | >5 | >5 | 4.60 ± 0.43 | 27.43 ± 1.22 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pulido, D.; Arranz-Trullén, J.; Prats-Ejarque, G.; Velázquez, D.; Torrent, M.; Moussaoui, M.; Boix, E. Insights into the Antimicrobial Mechanism of Action of Human RNase6: Structural Determinants for Bacterial Cell Agglutination and Membrane Permeation. Int. J. Mol. Sci. 2016, 17, 552. https://doi.org/10.3390/ijms17040552
Pulido D, Arranz-Trullén J, Prats-Ejarque G, Velázquez D, Torrent M, Moussaoui M, Boix E. Insights into the Antimicrobial Mechanism of Action of Human RNase6: Structural Determinants for Bacterial Cell Agglutination and Membrane Permeation. International Journal of Molecular Sciences. 2016; 17(4):552. https://doi.org/10.3390/ijms17040552
Chicago/Turabian StylePulido, David, Javier Arranz-Trullén, Guillem Prats-Ejarque, Diego Velázquez, Marc Torrent, Mohammed Moussaoui, and Ester Boix. 2016. "Insights into the Antimicrobial Mechanism of Action of Human RNase6: Structural Determinants for Bacterial Cell Agglutination and Membrane Permeation" International Journal of Molecular Sciences 17, no. 4: 552. https://doi.org/10.3390/ijms17040552