1. Introduction
Hepatocellular carcinoma is a primary malignancy of the liver. It accounts for the third leading cause of cancer deaths worldwide, with over 600,000 people affected [
1]. It mainly develops from chronic liver disease such as hepatitis B virus and hepatitis C virus infections. Emerging evidence has shown that there have been enormous changes of many signaling pathways during the development of liver cancer, with many surface proteins involved. These surface proteins have potential to become excellent therapeutic targets for liver cancer treatment.
The transmembrane-4 superfamily (TM4SF) is a group of cell-surface low molecular weight proteins that have four highly hydrophobic transmembrane domains.
TM4SF1 is an important member of the TM4SF. In particular, previous research showed that cervical cancer, lung cancer, squamous cell cancer, colon cancer, and breast cancer have elevated expression of
TM4SF1 mRNA, and that prostate cancer has elevated expression of
TM4SF1 protein [
2]. Our previous studies showed that 86% of patients with hepatitis B virus-related hepatocellular carcinoma have overexpressed
TM4SF1 in their liver cancer cells, but that adjacent normal tissues and normal liver tissues had no measureable expression of
TM4SF1 [
3]. Other studies reported that
TM4SF1 expression is closely related to the metastasis and recurrence of prostate cancer, non-small cell lung cancer, and breast cancer, and that
TM4SF1 expression is negatively associated with the survival of patients with squamous cell lung cancer [
4]. In addition, some members of the TM4SF family (
TM4SF3,
TM4SF5,
cD151, and
cD82) have roles in the invasion and metastasis of liver cancer [
5,
6,
7,
8]. However, few studies have investigated the role of
TM4SF1 in liver cancer. Thus, the purpose of the present study was to examine the role of
TM4SF1 in regulating the proliferation, migration, and invasion of liver cancer cells.
3. Discussion
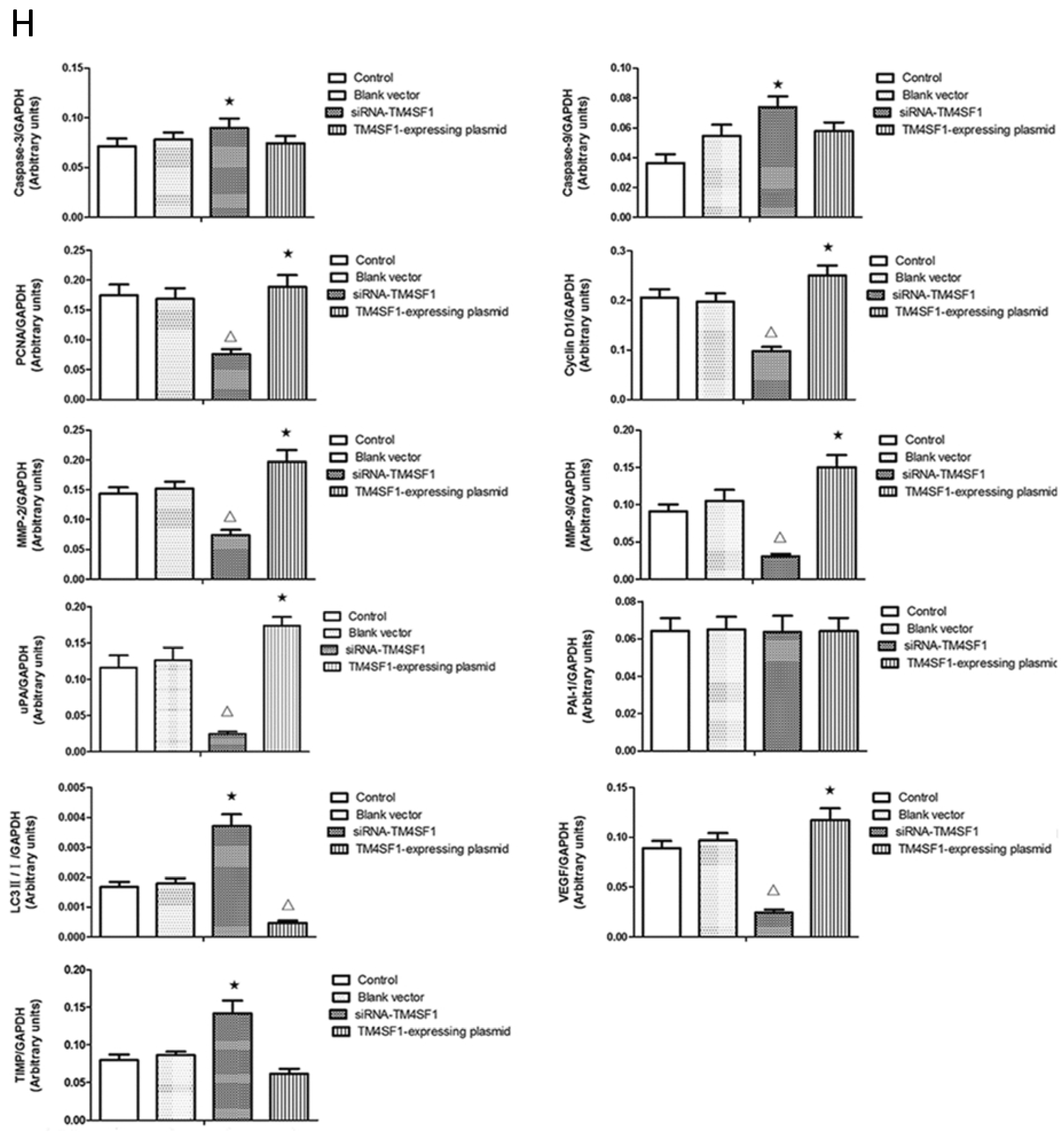
Cancer cells are characterized by increased proliferation and reduced apoptosis. Cyclin D1 promotes passage through phase G1 of the cell cycle and is overexpressed in liver cancer; overexpression of cyclin D1 appears to promote cancer cell invasion [
11]. In liver cancer patients, high proliferating cell nuclear antigen (
PCNA) expression is associated with increased involvement of blood vessels, and reduced postoperative disease-free survival time [
12]. The results of present study showed that cells with upregulated
TM4SF1 had increased expression of cyclin D1 and
PCNA and that silencing of
TM4SF1 reduced the expression of these two genes and also inhibited the proliferation and growth of HepG2 cells. This suggests that in the pathogenesis of liver cancer,
TM4SF1 upregulates cyclin D1 and
PCNA and thereby promotes the growth, proliferation, and invasion of cancer cells.
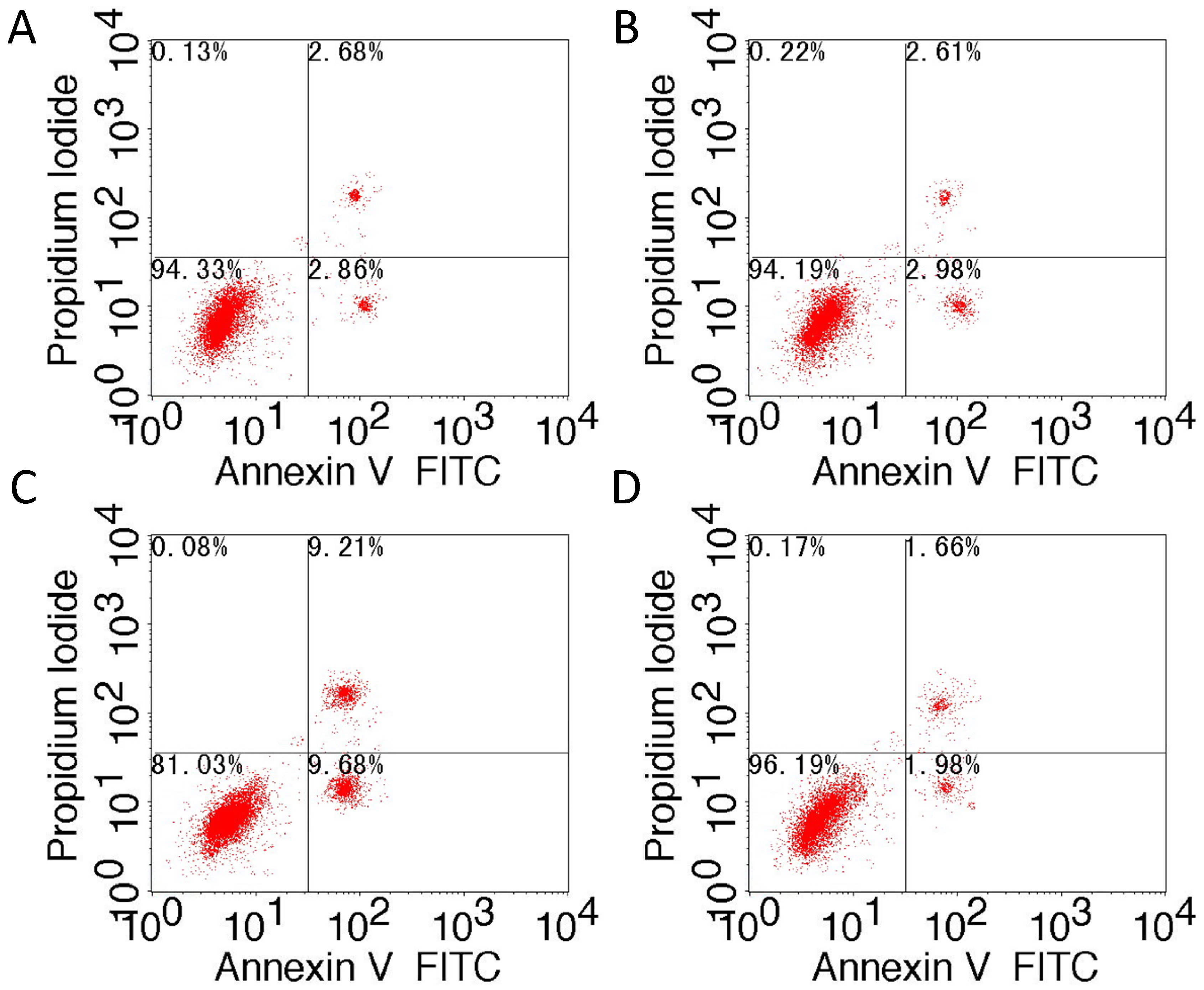
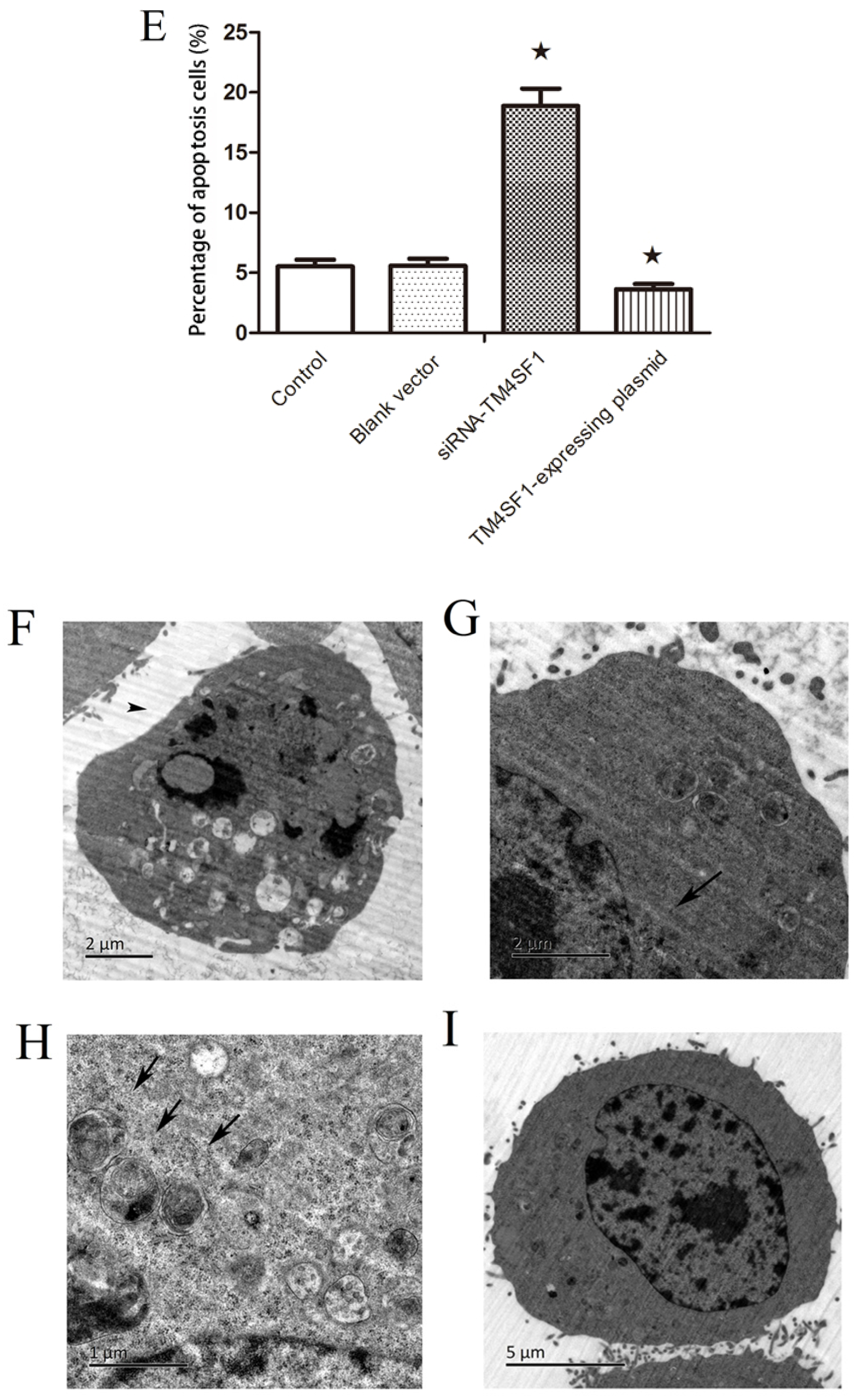
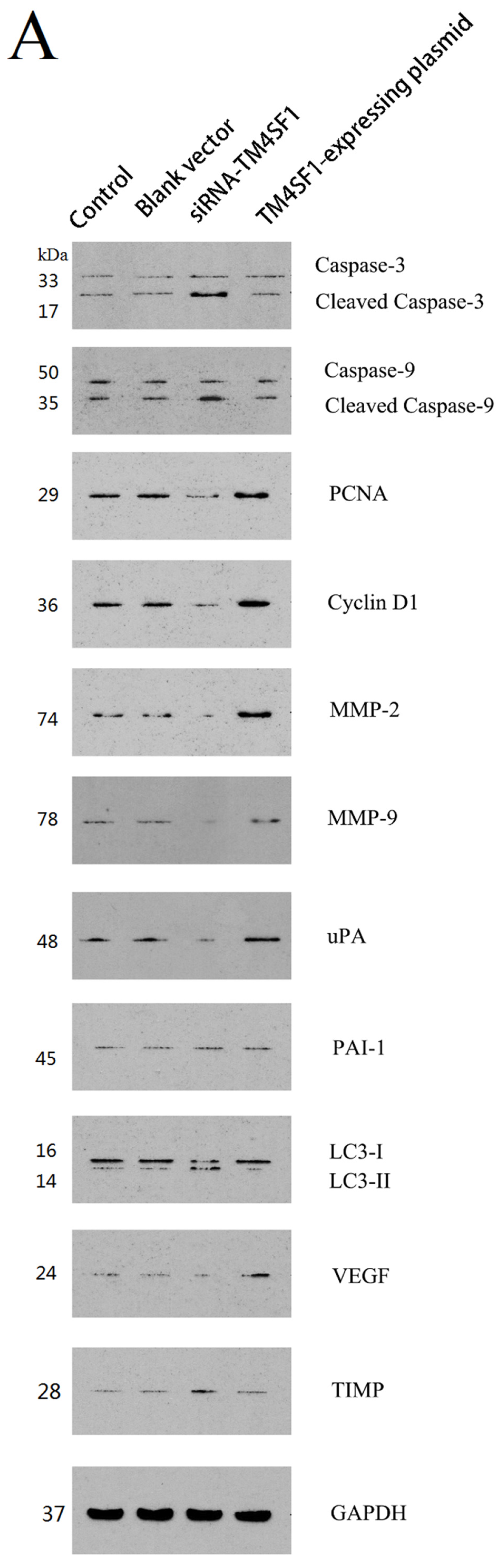
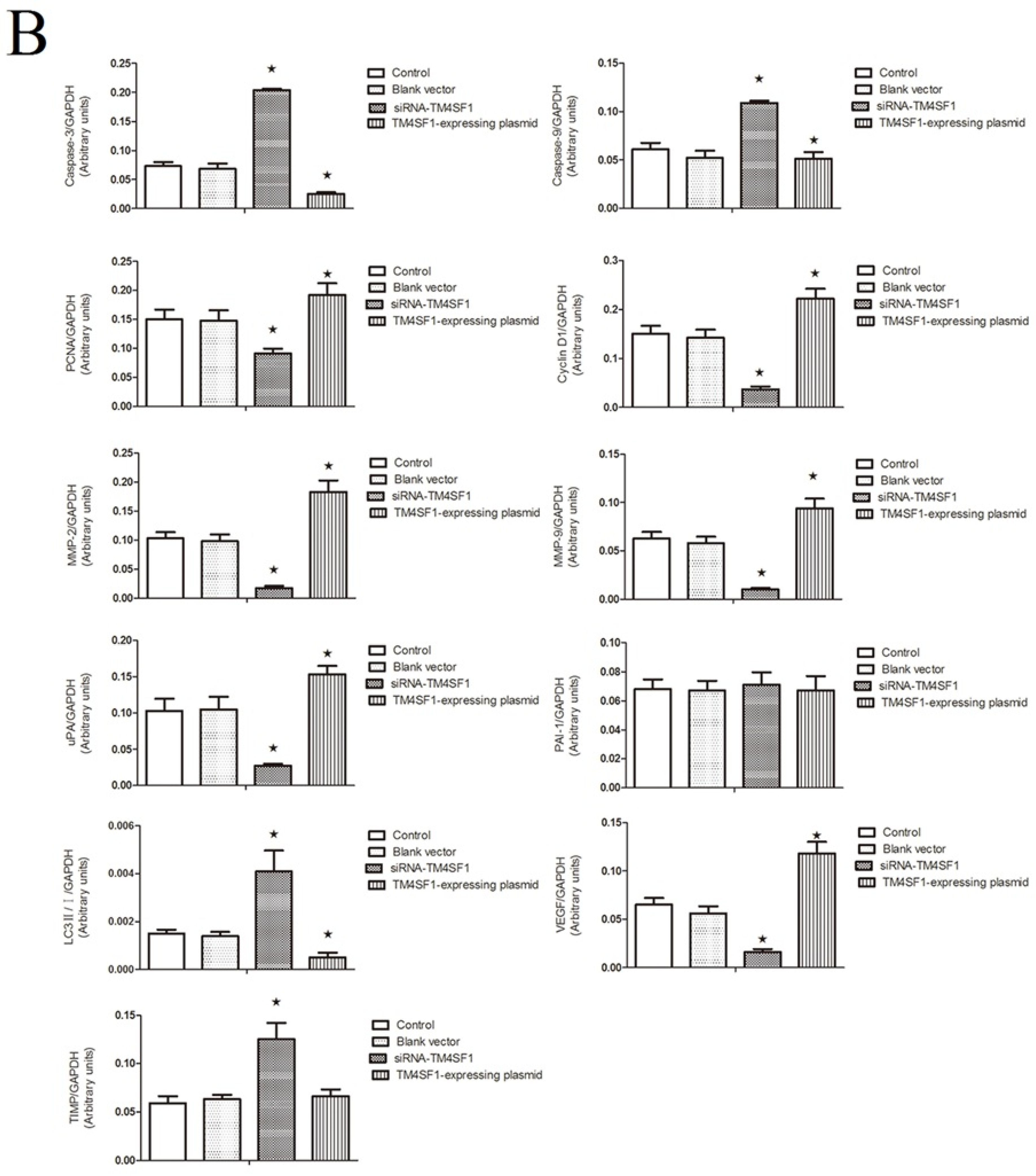
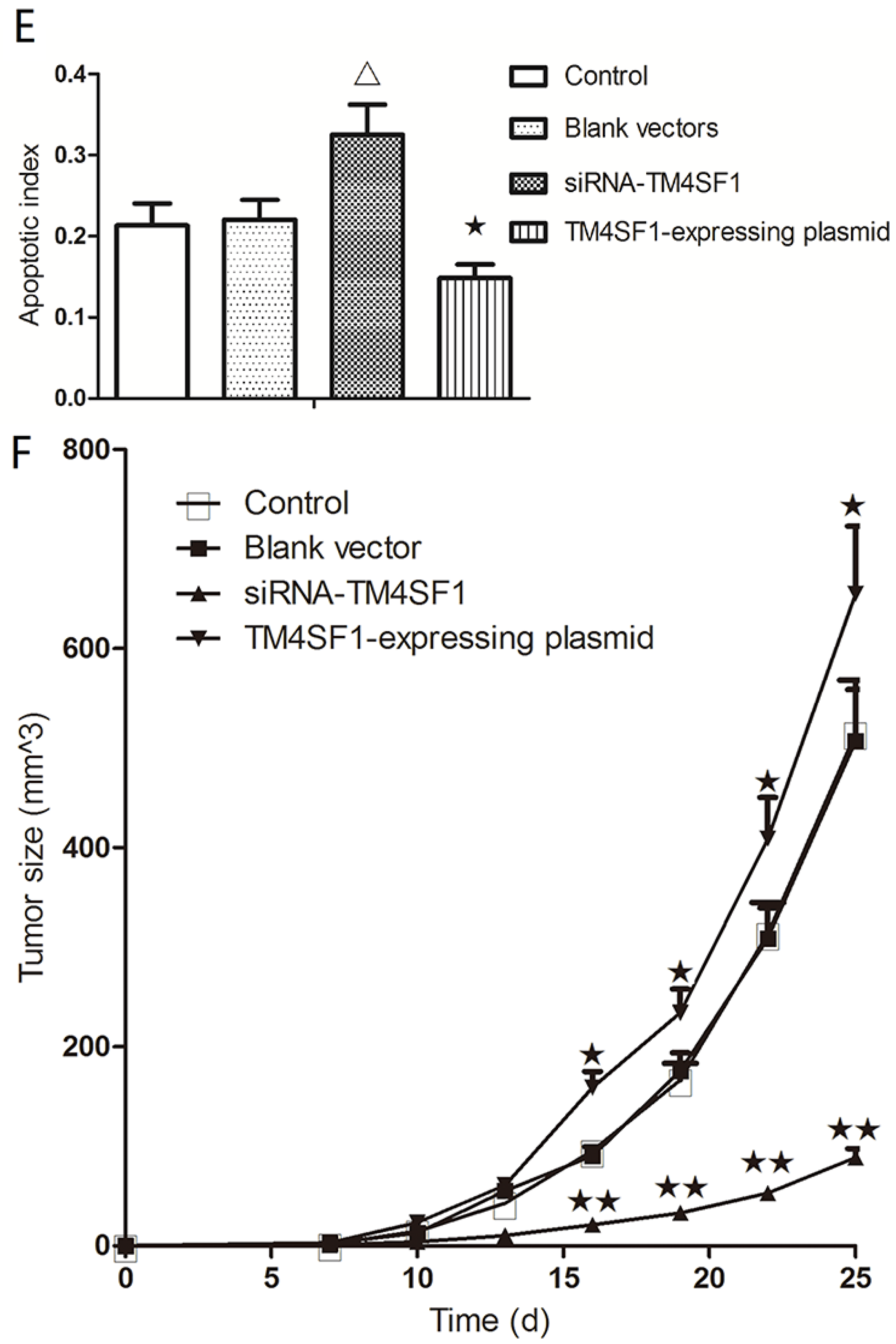
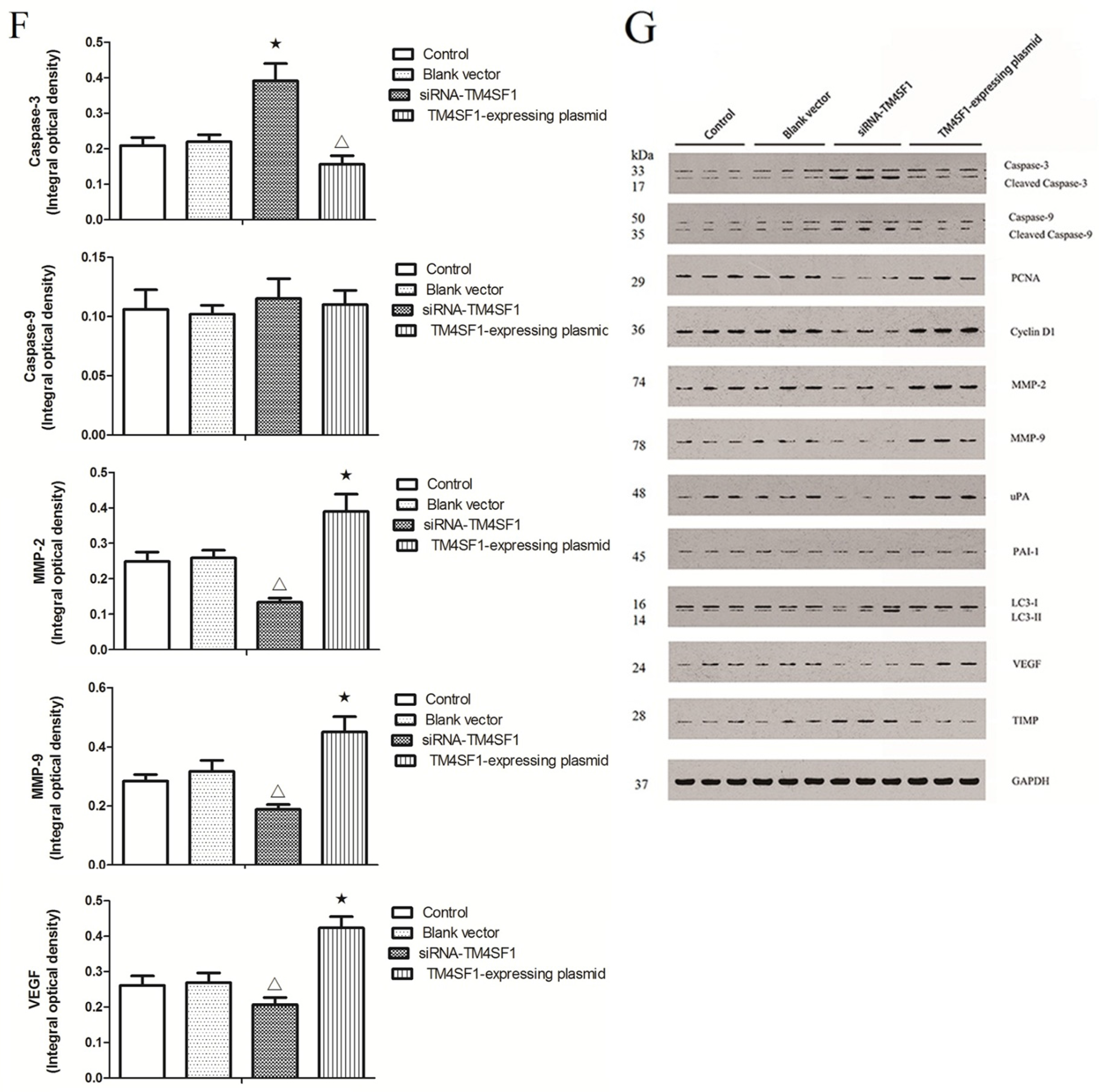
Our results showed that upregulation of TM4SF1 significantly inhibited the apoptosis of HepG2 cells, and that silencing of TM4SF1 expression with siRNA induced the apoptosis of these cells. We also found that upregulation of TM4SF1 inhibited the expression of caspase-3 and caspase-9 in HepG2 cells and growth of transplanted tumors and that silencing of TM4SF1 increased the expression of caspase-3 and caspase-9 in HepG2 cells and growth of transplanted tumors. Thus, we speculate that TM4SF1 upregulation inhibits apoptosis and induces abnormal proliferation of liver cancer cells by downregulating caspase-3 and caspase-9, and that this leads to the onset and progression of liver cancer.
Recent studies indicate that autophagy may inhibit the onset and progression of numerous cancers. For example, cells with stable transfection of
beclin-1 had increased autophagy and reduced tumorigenesis [
13]. Downregulation of beclin-1 reduces cell autophagy and prolongs the life cancer cells, leading to increased development of cancers [
14,
15]. Our results showed that transfection of HepG2 cells with
TM4SF1-expressing plasmids significantly increased
TM4SF1 expression, and that this markedly inhibited the autophagy of HepG2 cells, as indicated by the presence of fewer autophagosomes and reduced LC3II expression. Silencing of
TM4SF1 increased the autophagy of HepG2 cells, and this was accompanied by significant increases in the number of autophagosomes and expression of LC3II. Thus, inhibition of the autophagy of cancer cells may be one of the mechanisms underlying
TM4SF1-induced tumorigenesis.
Although autophagy can inhibit tumorigenesis, other studies showed that autophagy can also promote the survival of cancer cells. For example, when tumor growth overwhelms angiogenesis, there may be focal ischemia and hypoxia; under these stressful conditions, which often occur at the center of cancers where new blood vessels do not form, the survival of cancer cells depends on catabolism during autophagy [
16]. The present study of nude mice was a preliminary examination of the role of autophagy in the early phase of tumorigenesis (25 days after subcutaneous injection of HepG2 cells). The results showed that upregulation of
TM4SF1 inhibited the autophagy of liver cancer cells and increased the susceptibility to tumorigenesis, and that downregulation of
TM4SF1 markedly promoted the autophagy of cancer cells and decreased the susceptibility to tumorigenesis. Thus, we speculate that in the early phase of liver cancer, downregulation of
TM4SF1 plays an important role in the promotion of tumorigenesis.
Some findings suggest that there is crosstalk between autophagy and apoptosis, and that
caspase-3 and
caspase-9 may mediate this effect.
Caspase-9 may form complexes with ATG7 and induce the formation of LC3-II and thereby promote autophagy [
17].
Caspase-3 may be a molecular switch that mediates the crosstalk between autophagy and apoptosis, and activated
caspase-3 can promote secretion of autophagic vacuoles [
18]. Our results showed that upregulation of
TM4SF1 downregulates the expression of
caspase-3 and
caspase-9, and inhibits the apoptosis and autophagy of HepG2 cells, thereby promoting cell proliferation and facilitating tumorigenesis. On the contrary, downregulation of
TM4SF1 upregulates the expression of
caspase-3 and
caspase-9, and activates apoptosis and autophagy of HepG2 cells, thereby suppressing cell proliferation and tumorigenesis. Thus, we speculate that in the early pathogenesis of liver cancer,
TM4SF1 has a central role in the inhibition of apoptosis and autophagy that is mediated through its effects on
caspase-3 and
caspase-9.
Vascular endothelial growth factor (
VEGF) is a highly specific mitogen of the vascular endothelium that increases the permeability of microvessels, blocks the degeneration of newly generated blood vessels, increases glucose transportation by the vascular endothelium, promotes the division and proliferation of the vascular endothelium, and facilitates the migration of endothelial cells [
19,
20,
21]. There is evidence that
VEGF promotes the secretion of some enzymes that facilitate the metastasis of cancers, and that overexpression of
VEGF can induce
MMP-2 and
MMP-9, which may be a major mechanism underlying the invasion and metastasis of highly invasive cancers [
22,
23].
MMP-9 disrupts the basement barrier and promotes the migration of capillary endothelial cells to initiate cancer angiogenesis [
24]. The
MMP-2 (located at 16q21) is a major component of the MMP family and has extensive distribution. This protein degrades the ECM and thereby promotes the migration of cancer cells across the ECM and basement barrier and the subsequent metastasis of cancer cells through connective tissues [
25,
26,
27,
28]. The substrates of
MMP-2 and
MMP-9 are mainly the skeletal components of the basement membrane, such as type IV and type V collagen.
TIMP can irreversibly bind to MMP, inhibit MMP activity, block degradation of the ECM, and thereby inhibit the invasion and metastasis of cancers [
29]. The results of our studies of HepG2 cells and transplanted tumors showed that
TM4SF1 promoted the expression of
uPA,
MMP-2, and
MMP-9, and that silencing of
TM4SF1 inhibited the expression of
uPA,
MMP-2, and
MMP-9 and elevated
TIMP expression. Our results also indicated that
TM4SF1 had no effect on the expression of
PAI-1. We speculate that
TM4SF1-mediated upregulation of
uPA,
MMP-2, and
MMP-9 increases the degradation of ECM by cancer cells, leading to invasion and metastasis of cancer cells. Silencing of
TM4SF1 appears to restore the balance between MMP and
TIMP, increase the inhibition of MMP by
TIMP, reduce degradation of the ECM, thus inhibiting the invasion and metastasis of cancer cells.
TM4SF1 may promote the invasion and metastasis of cancers through one or more mechanisms. First, it may promote the vascular endothelial cells in the cancer to initiate angiogenesis, which indirectly promotes cancer cell invasion and metastasis. On the other hand, it may strengthen the interaction between cancer cells and the ECM, which facilitates the invasion and metastasis of cancer cells [
30]. In addition,
TM4SF1 overexpression is also involved in the formation of pseudopodia in cancer cells, and this facilitates the invasion and metastasis of cancer cells [
31,
32], and
TM4SF1 has recently been reported to stimulate breast cancer cell invasion and migration through PI3K/AKT/mTOR pathway [
33]. It should be noted that
TM4SF1 is highly expressed in liver cancer, and liver cancer patients with
TM4SF1 overexpression have worse five-year survival rates than those with low
TM4SF1 expression [
34], supporting our results.
TM4SF1 expression is also elevated in lung cancer, pancreatic cancer, liver cancer, and cervical cancer, leading to its classification as a tumor-associated antigen [
35,
36,
37,
38]. After injection of a human-mouse chimeric monoclonal antibody against
TM4SF1, 22.2% (4/18) of patients with breast cancer, colon cancer, or non-small cell lung cancer produced antibodies against antibody, and these aggregated around cancer cells [
39]. Our results confirmed that
TM4SF1 was closely related to the migration and invasion of HepG2 cells. Overexpression of
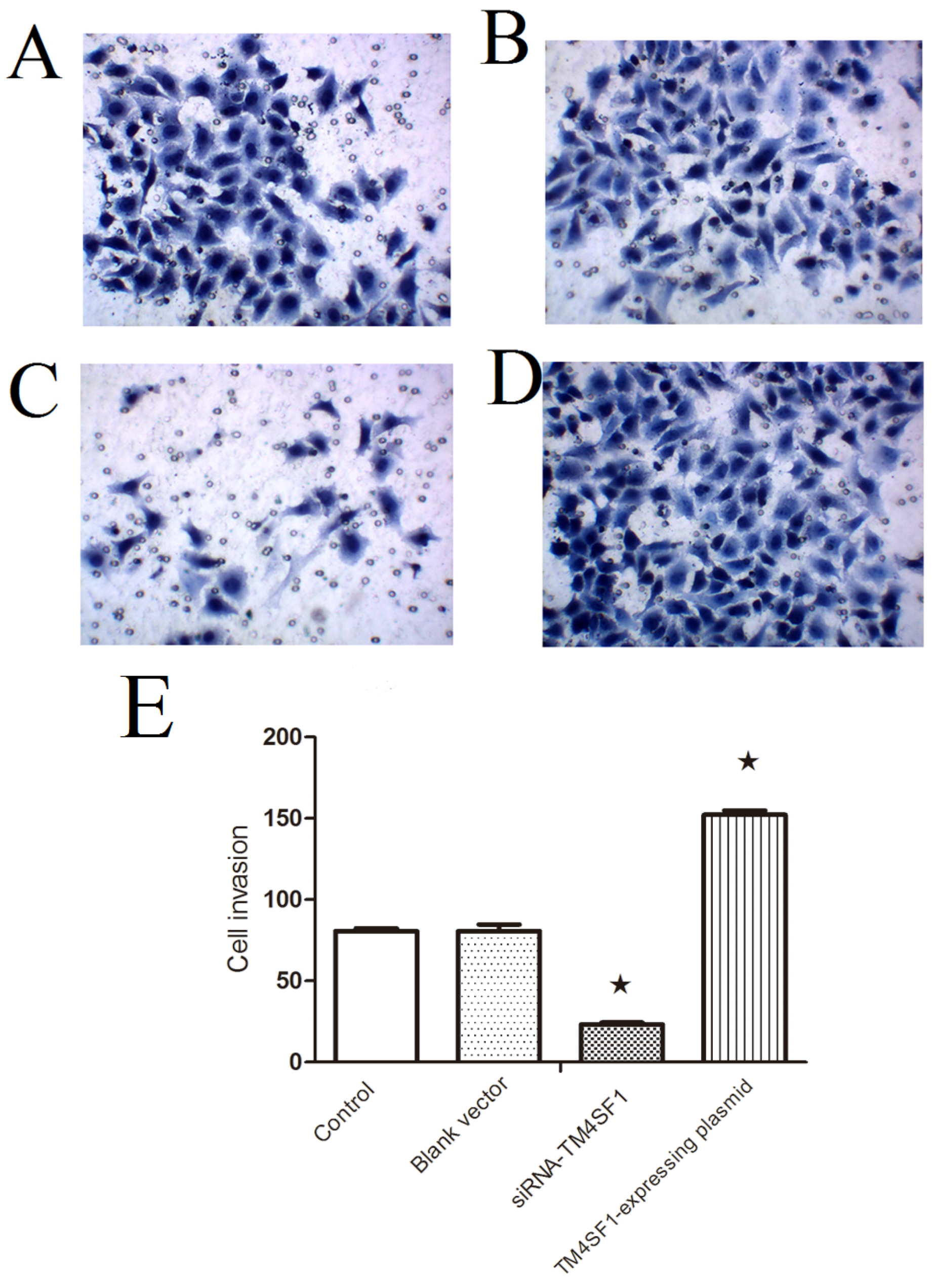
TM4SF1 in HepG2 cells significantly increased the migration of cells across the Matrigel membrane and promoted the growth of transplanted tumors. Silencing of TM4SFl markedly reduced the migration of HepG2 cells across the Matrigel membrane and inhibited the growth of transplanted tumors. This suggests that silencing of TM4SFl expression should be considered as a potential new strategy for the therapy of liver cancer.
4. Experimental Section
4.1. Materials
Human liver cancer cells (HepG2 cells) were provided by the Department of Infectious Diseases of the Affiliated Xiangya Hospital of Central South University. TM4SF1-expressing plasmids were prepared by Shanghai Genepharma Co., Ltd. (Shanghai, China). Lipofectamine® 2000 and the Trizol reagent were from Invitrogen (Carlsbad, CA, USA); RPMI-1640, trypsin, fetal bovine serum (FBS), and G418 were from Gibco (Grand Island, NY, USA); monoclonal antibodies against TM4SF1, MMP-2, PAI-1, uPA, TIMP, and PCNA were from Abcam (Cambridge, UK); monoclonal antibodies against caspase-9 and caspase-3 were from Bioss (Woburn, MA, USA); monoclonal antibodies against LC3I/II and cyclin D1 were from Cell Signaling Technology (Danvers, MA,USA); monoclonal antibodies against MMP-9 were from ProteinTech Group (Chicago, IL, USA); monoclonal antibodies against GAPDH were from Santa Cruz Biotechnology (Dallas, TX, USA); Transwell assays and matrigel were from BD Biosciences (San Jose, CA, USA); ultraSensitive TM-SP was from Fuzhou Maxim Biotech Co., Ltd. (Fuzhou, China); and ECL+ was from Amersham (Piscataway, NJ, USA).
4.2. Plasmid Construction
The open reading frames (ORFs) of hTM4SF1 were cloned from HEK293 cell cDNA using the primer pairs hTM4SF1-F and hTM4SF1-R, and hTM4SF1 based on the hTM4SF1 (GenBank accession no. 4071 and Refseq: NM_014220) sequences. The primer pairs for TM4SF1 were 5′-ATGTGCTATGGGAAGTGTGCAC-3′ (forward), and 5′-TGGTTGTCGTTATACTGACGATT-3′ (reverse). pGBKT7-hTM4SF1 was constructed by cloning hTM4SF1 into the expression vector pGBKT7 (Clontech, California, CA, USA), which encoded the full-length TM4SF1 fused to the GAL4 DNA-binding domain for yeast two-hybrid screening. pcDNA3.1-Myc-hTM4SF1 was obtained by the respective cloning of hTM4SF1 genes into the mammalian expression vector pcDNA3.1-Myc (Invitrogen, California, CA, USA) to express hTM4SF1 fused to a N-terminal Myc epitope tag. pEGFP-N1 empty vector (Clontech) encoding enhanced green fluorescent protein (EGFP), and was used as a control. pGEM-hTM4SF1 was constructed by cloning hTM4SF1 into the transcription vector pGEM-4Z (Promega, Madison, AL, USA).
4.3. Cell Culture and Plasmid Transfection
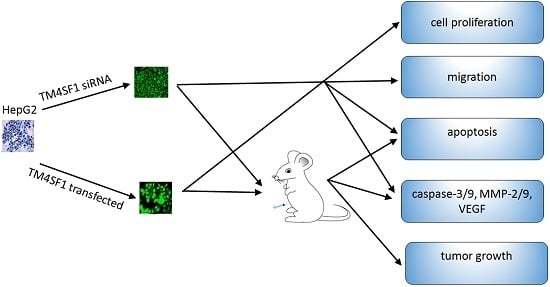
HepG2 cells were maintained in RPMI1640 medium that contained 100 mL/L FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified environment with 5% CO2. Opti-MEM® I was used to dilute blank vectors, TM4SF1-expressing plasmids, and Lipofectamine® 2000, followed by incubation at room temperature for 5 min. The diluted blank vectors and TM4SF1-expressing plasmid solutions were independently mixed with Lipofectamine® 2000, followed by incubation at room temperature for 20 min. The resultant solution was transferred into plates containing HepG2 cells, followed by incubation for 72 h. Cells were harvested for real-time PCR, flow cytometry, transmission electron microscopy, Transwell migration assay, MTT assay, Western blotting, and subcutaneous injection into Foxn1−/− nude mice.
4.4. Gene Silencing with siRNA
Three pairs of siRNAs that targeted TM4SF1 were designed and synthesized: (i) siRNA-TM4SF1-497: 5′- GCGAUGCUUUCUUCUGUAUTT-3′ (forward), 5′-AUACAGAAGAAAGCAUCGCTT-3′ (reverse); (ii) siRNA-TM4SF1-733: 5′-GGCUCUUGGUGGAAUUGAATT-3′ (forward), 5′-UUCAAUUCCACCAAGAGCCTT-3′ (reverse); and (iii) siRNA-TM4SF1-813: 5′-GCUCUCACCAACAGCAAUATT-3′ (forward), 5′- UAUUGCUGUUGGUGAGAGCTT-3′ (reverse). Scrambled siRNA (forward: 5′-UUCUCCGAACGUGUCACGUTT-3′; reverse: 5′-ACGUGACACGUUCGGAGAATT-3′) was synthesized as a control. Opti-MEM® I was used to dilute these siRNAs or Lipofectamine® 2000, followed by incubation at room temperature for 5 min. The resultant siRNA was mixed with Lipofectamine® 2000, incubated at room temperature for 20 min, and then transferred onto plates containing HepG2 cells. Cells were maintained for 24 h and then harvested for real-time PCR, flow cytometry, transmission electron microscopy, Transwell migration assay, MTT assay, Western blotting, and subcutaneous injection into Foxn1−/− nude mice (see below).
4.5. Real-Time PCR
Total RNA was extracted from HepG2 cells transfected with siRNA-TM4SF1, TM4SF1-expressing plasmids, and blank vectors and from HepG2 cells without transfection by use of the Trizol reagent according to manufacturer’s instructions. The MyIQ real-time PCR system (Bio-Rad, Hercules, CA, USA) was used to measure mRNA expression of β-actin (housekeeping gene) and TM4SF1. The primer for β-actin was 5′-CATTAAGGAGAAGCTGTGCT-3′ (forward), 5′-GTTGAAGGTAGTTTCGTGGA-3′ (reverse) and the primer for TM4SF1 was 5′-AAGGGGGAGAAAACCTAGCA-3′ (forward), 5′-CCAGCCCAATGAAGACAAAT-3′ (reverse).
4.6. Flow Cytometry
HepG2 cells that were transfected with siRNA-TM4SF1, TM4SF1-expressing plasmids, and blank vectors and HepG2 cells without transfection were subjected to Annexin V-FITC/PI staining according to the manufacturer’s instructions. After washing in PBS, cells were re-suspended in binding buffer and cell density was adjusted to 5 × 105/mL. Then, 195 μL of cell suspension was mixed with 5 μL of Annexin V-FITC, followed by incubation at room temperature for 10 min. After one wash in PBS, cells were re-suspended in 190 μL of binding buffer, followed by addition of 10 μL of 20 µg/mL propidium iodide. Finally, cells were washed and then analyzed using a flow cytometer. Percentage of apototosis cells (%) = (number of Annexin V+PI− and Annexin V+PI+ cells)/total cells × 100%.
4.7. Transmission Electron Microscopic Examination
The morphology of HepG2 cells that were transfected with siRNA-TM4SF1, TM4SF1-expressing plasmids, and blank vectors and HepG2 cells without transfection were examined using a transmission electron microscope at the Xiangya School of Medicine Electron Microscope Facility, Central South University, China. The cells were fixed in phosphate-buffered 2.5% glutaraldehyde for 24 h, postfixed in phosphate-buffered 2% osmium tetroxide for 2 h, dehydrated in ascending concentrations of acetone, infiltrated over 24 h with Spurr’s resin, and observed using a Hitachi-7700 transmission electron microscope (Ibaraki, Japan).
4.8. Transwell Migration Assay
In the Transwell migration assay, HepG2 cells transfected with siRNA-TM4SF1, TM4SF1-expressing plasmid, or blank vectors and HepG2 cells without transfection were seeded into the upper Transwell chambers (5 × 104 cells) and maintained in serum free medium. In the lower chamber, medium containing 150 mL/L FBS was added, followed by incubation at 37 °C with 5% CO2 for 24 h. The upper chambers were taken out and the inner cells were removed from the upper chambers, which was then washed twice and fixed in 95% ethanol, followed by hematoxylin staining. Cells were observed under an inverted microscope. Five fields were randomly selected and positive cells were classified as invasive.
4.9. Animal Study
Foxn1−/− nude mice (6 to 8 weeks, Department of Animal Experiments, Central South University) were used in all animal studies. National Institutes of Health Guidelines for Care and Use of Laboratory Animals were observed. HepG2 cells transfected with siRNA-TM4SF1, TM4SF1 expressing plasmid, or blank vector and HepG2 cells without transfection were subcutaneously inoculated into Foxn1−/− nude mice (1 × 106 HepG2 cells/mouse). The tumor volume was measured as maximum longest diameter × minimum shortest diameter2 × 0.52. At 25 days after subcutaneous injection, mice were sacrificed and the transplanted tumors were collected for analysis. All studies were approved by the Institutional Review Board of Third Xiangya Hospital, Central South University, China (4 March 2015, No: 2015-S035).
4.10. Western Blotting
HepG2 cells transfected with siRNA-TM4SF1, TM4SF1-expressing plasmids, blank vectors and cells without transfection, and the transplanted tumors of nude mice were harvested. Total protein was extracted from cells and tissues for measurement of protein expression. Cell extracts were prepared using a lysis buffer containing 20 mM HEPES (pH 7.4), 0.5% Triton X-100, 150 mM NaCl, 12.5 mM β-glycerophosphate, 50 mM NaF, 1 mM DTT, 1 mM sodium orthovanadate, 2 mM EDTA, 1 mM PMSF, and protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA). Protein concentration of cell extracts was determined by the Bradford protein reagent (Bio-Rad), using BSA as a standard. Equal amounts of cell extracts were resuspended in Laemmli loading buffer (Bio-Rad), boiled and subjected to SDS-polyacrylamide gel electrophoresis to separate proteins on 4%–20% polyacrylamide minigels (Invitrogen). Proteins were electrotransferred to Immobilon-P membranes (Millipore, Billerica, MA, USA), membranes were blocked with Tris-buffered solution (TBS) with 0.1% Tween 20 (TBS-T), containing 5% non-fat dry milk (Bio-Rad), and probed for 20 h at 4 °C with the corresponding primary antibodies, such as the monoclonal antibody against TM4SF1, caspase-3, caspase-9, PCNA, cyclin D1, MMP-2, MMP-9, uPA, PAI-1, LC3, VEGF, TIMP and GAPDH. After washing five times in TBS-T, membranes were incubated with the corresponding anti-rabbit, anti-mouse, or anti-goat secondary IgG-HRP conjugates diluted at 1:12,000 in TBS-T/5% milk, before being washed three times; bands were revealed by incubation with enhanced chemiluminescence reagents (ECL+) followed by exposure to X-ray films. Densitometric analysis of intensities of the protein bands from three independent experiments was performed using the Quantity One software (Bio-Rad). Levels of TM4SF1, caspase-3, caspase-9, PCNA, cyclin D1, MMP-2, MMP-9, uPA, PAI-1, LC3, VEGF, and TIMP were normalized to those of GAPDH, and data were expressed as arbitrary units.
4.11. Terminal dUTP Nick End-Labeling (TUNEL) Staining
HepG2 cells transfected with siRNA-TM4SF1, TM4SF1-expressing plasmids, blank vectors and cells without transfection, and transplanted tumors of nude mice were harvested and processed for the measurement of apoptosis. The terminal dUTP nick end-labeling (TUNEL) assay was performed using the TdT-FragEL TM DNA Fragmentation Detection kit (Calbiochem/Oncogene Research Products, Cambridge, MA, USA) according to the manufacturer’s instructions. Briefly, 4 μm sections from the paraffin-embedded samples were dewaxed with xylene and hydrated using graded alcohols, and the specimens were treated with 20 mg/mL proteinase K for 5 min and with 0.6% H2O2 in methanol to eliminate endogenous peroxidase activity. Afterward, the sections were treated with the TDT enzyme and immersed in a biotinylated nucleotides solution. Apoptotic cells were detected using streptavidin–peroxidase conjugate followed by diaminobenzidine staining. All sections were observed under the same magnification, light source, brightness, color saturation, gain, and contrast. Five fields were randomly selected from each section, and images were processed with Motic Fluo 1.0 image analysis software (Motic China Group Co., Ltd., Guangzhou, China). TUNEL-positive cells had yellow-brown granules in the nucleus. After adjusting optical density, the apoptotic index was calculated by division of the number of labeled cells by the total number of cells in six high power fields (original magnification: 400×).
4.12. Immunohistochemistry
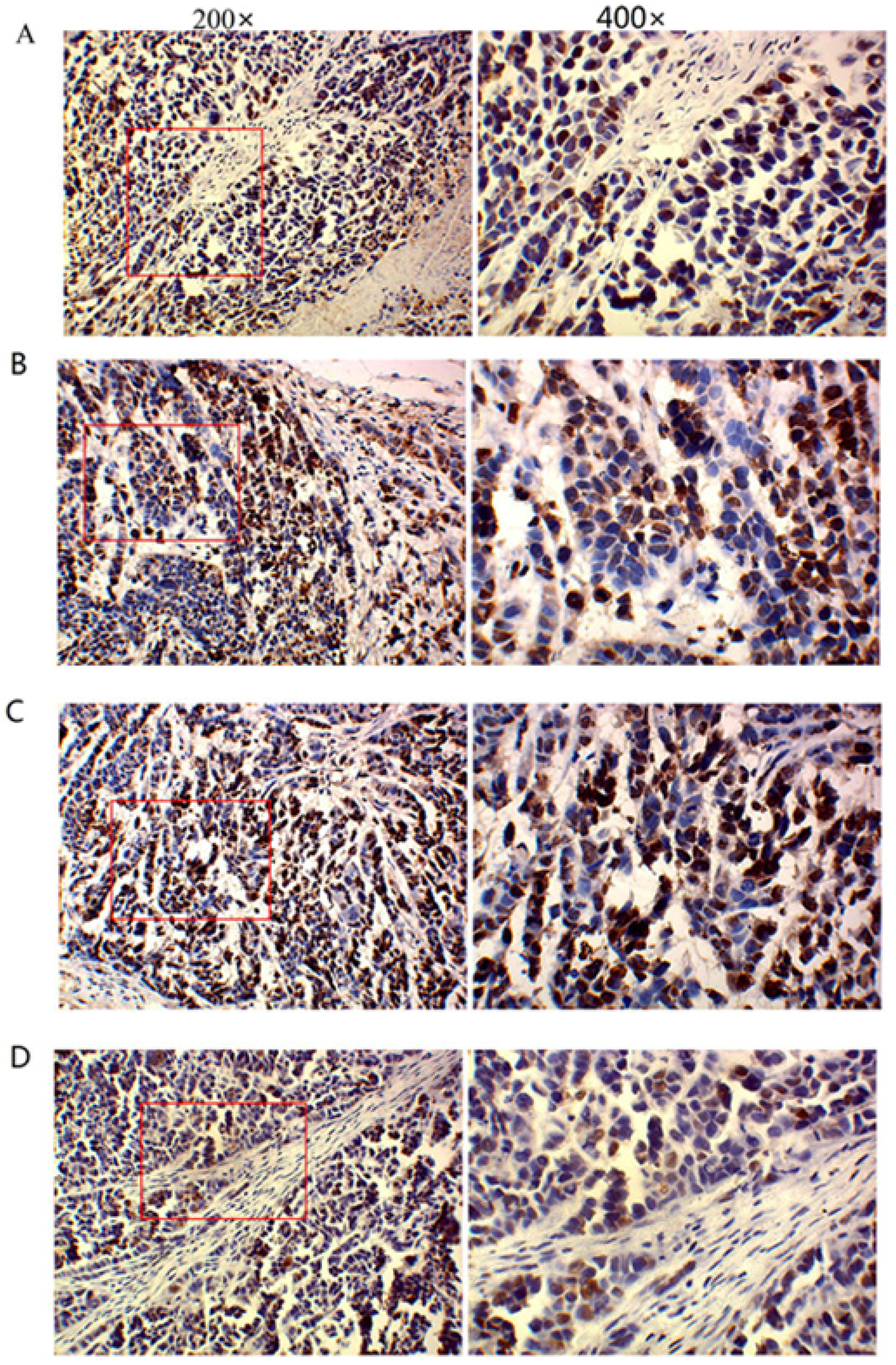
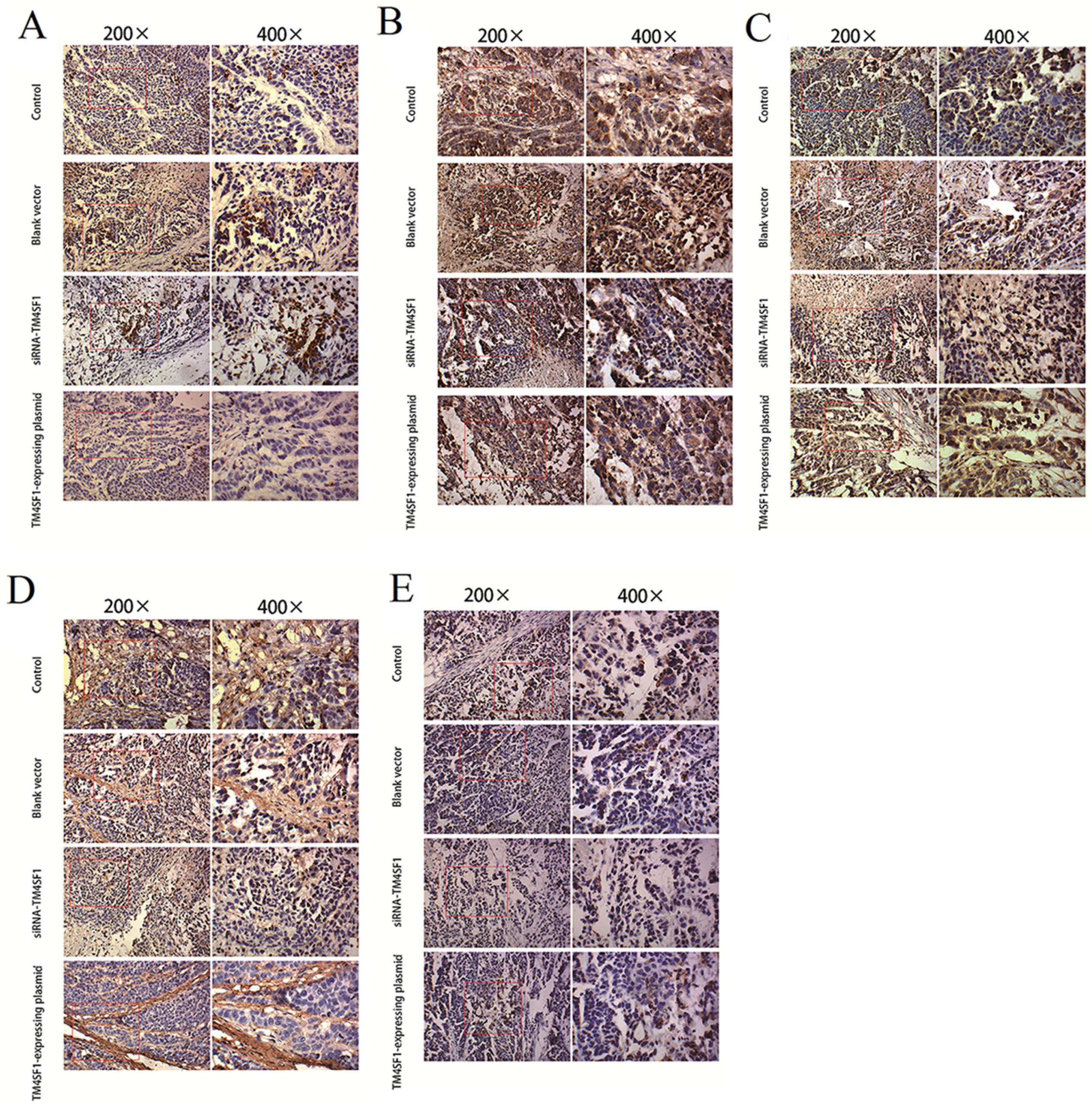
Immunohistochemistry was performed according to manufacturer’s instructions by use of the SP method (UltraSensitive TM-SP). In brief, sections were routinely deparaffinized and treated with methanol that contained 3% hydrogen peroxide for 30 min. Then, sections were blocked in normal serum (50 μL/section) for 10 min at room temperature, and incubated with the primary antibody (caspase-3, caspase-9, MMP-2, MMP-9 and VEGF) or PBS (negative control) at 4 °C overnight. After incubation with biotin-conjugated secondary antibody (50 μL/section) for 10 min at room temperature, 50 μL of streptavidin-peroxidase was added, followed by incubation at room temperature for 10 min. Sections were then treated with freshly prepared DAB solution for 3−10 min depending on the staining intensity (observed by microscopy). Sections were washed three times in PBS (3 min each) between steps. After staining, sections were counterstained with hematoxylin for 2 min and then treated with ethanol in HCl. Sections were washed in water, dehydrated in ethanol, transparentized in xylene, mounted with neutral gum, and observed under a light microscope. Positive cells had yellow-brown granules in the cytoplasm. Endothelial cells with yellow-brown granules in the cytoplasm were VEGF-positive cells; non-endothelial cells with yellow-brown granules in the cytoplasm were caspase-9- or MMP-2-positive cells, and monocytes with yellow-brown granules were positive for caspase-3. The IOD values for caspase-3, caspase-9, MMP-2, MMP-9, and VEGF cells were determined independently.
4.13. Statistical Analysis
Statistical analysis was performed using the GraphPad Prism 5 program for Windows (Graphpad Software, San Diego, CA, USA). Statistical differences between experimental groups were evaluated by a one-way ANOVA with repeated measures, followed by post hoc comparisons with Tukey’s multiple paired comparison test. Values are expressed as mean ± SD. A p ≤ 0.05 was considered significant.
5. Conclusions
Taken together, our results showed that overexpression of
TM4SF1 significantly increased the proliferation and tumorigenesis of liver cancer cells. Moreover, upregulation of
TM4SF1 downregulates the expression of pro-apoptotic genes (
caspase-3 and
caspase-9), upregulates the expression of genes related to cell proliferation and cell cycle progression (cyclin D1 and
PCNA), inhibits cell apoptosis and autophagy, and increases cell proliferation. In addition, when cancer cells with
TM4SF1 overexpression were injected into nude mice, this increased the expression of genes related to angiogenesis (
uPA,
MMP-2,
MMP-9 and
VEGF), decreased the expression of
TIMP (an inhibitor of MMP), and led to promotion of angiogenesis and tumor growth. Based on these findings,
TM4SF1 appears to enhance the invasion of cancer cells by several mechanisms (
Figure S4). Silencing of
TM4SF1 expression downregulates the expression of genes related to regulation of the cell cycle and cell proliferation (cyclin D1 and
PCNA), upregulates the expression of genes related to apoptosis and autophagy (
caspase-3 and
caspase-9). Silencing of
TM4SF1 also increases the expression of
TIMP (an inhibitor of MMP), inhibits the expression of pro-angiogenic genes (
uPA,
MMP-2,
MMP-9, and
VEGF) and suppresses the proliferation, invasion and metastasis of cancer cells. Thus, inhibition of
TM4SF1 expression may be a useful strategy to inhibit tumor growth and to reduce the migration and invasion of cancer cells.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









