
Mechanism of Mitochondrial Connexin43′s Protection of the Neurovascular Unit under Acute Cerebral Ischemia-Reperfusion Injury

 ,
,
Abstract
:
1. Introduction
2. Results
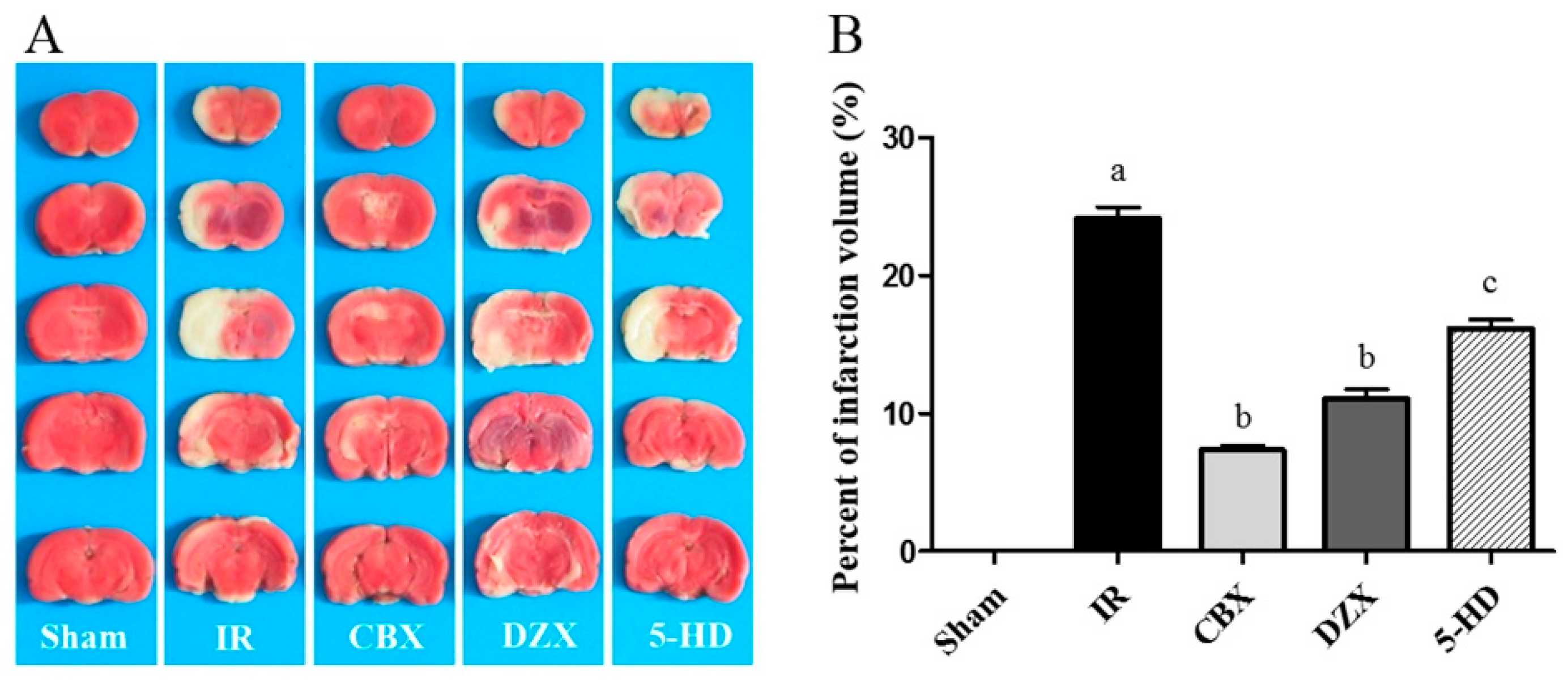
2.1. Cerebral Infarct Volume after MCAO in Different Groups
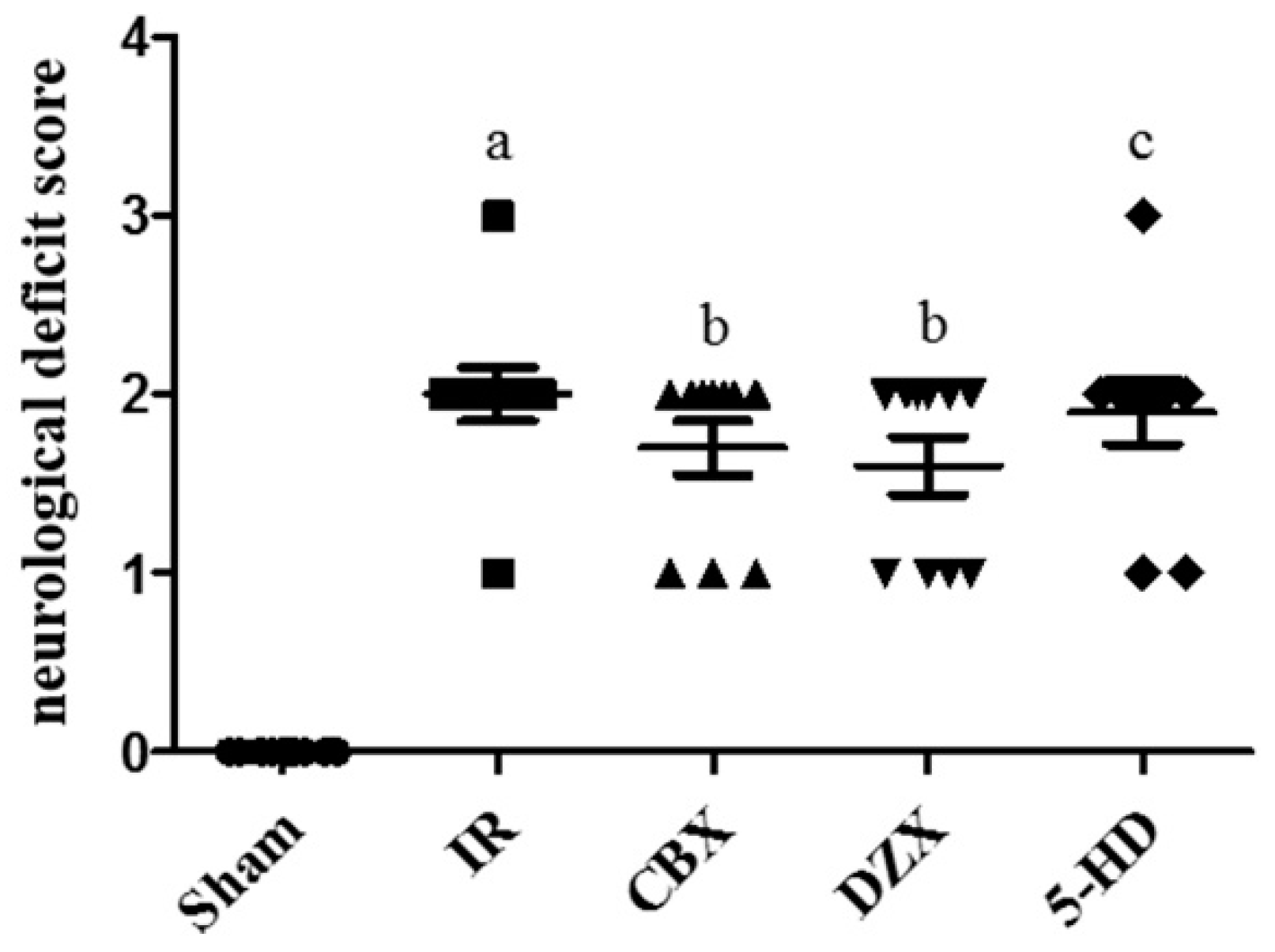
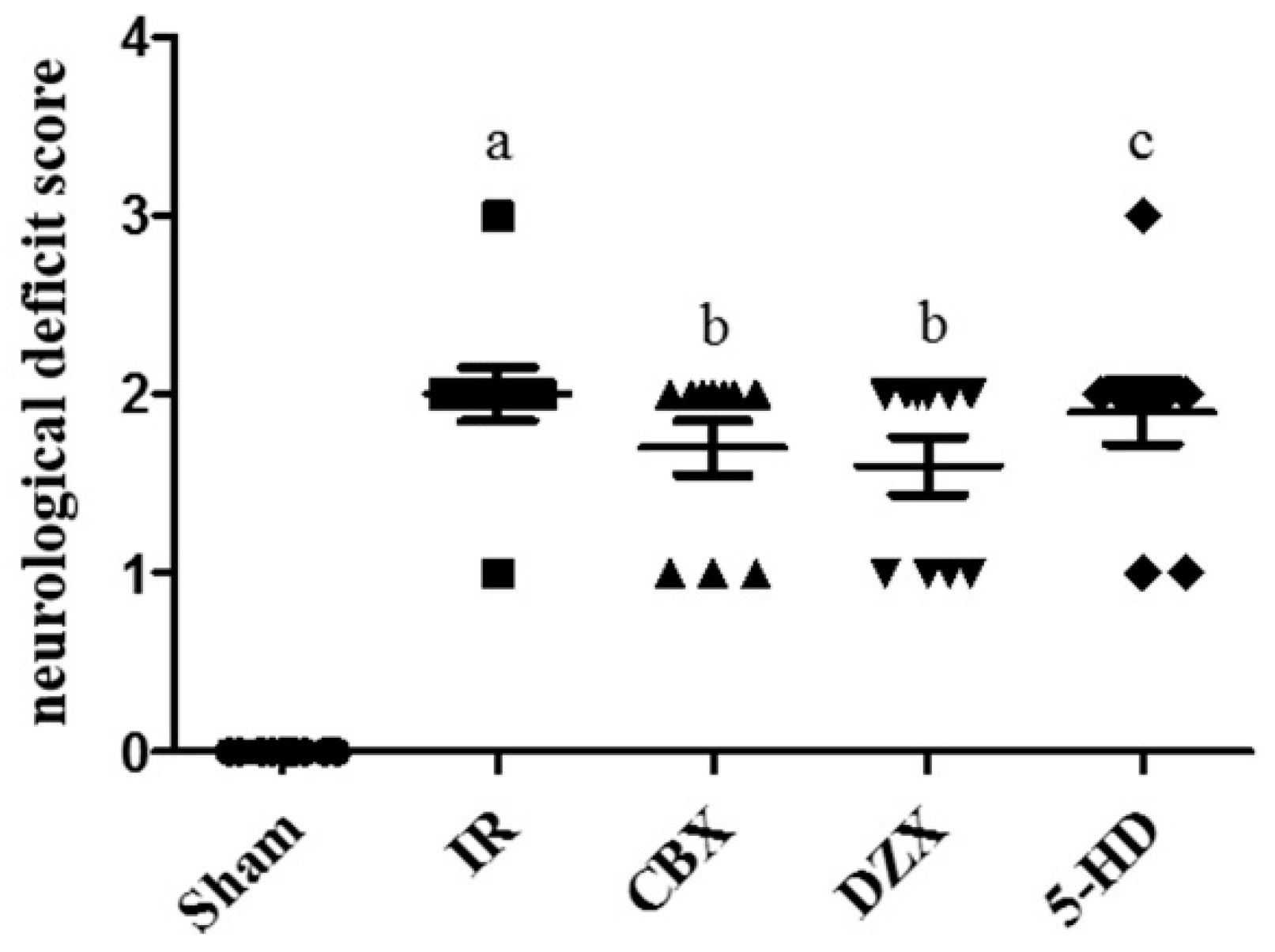
2.2. Neurological Deficit Scores after MCAO
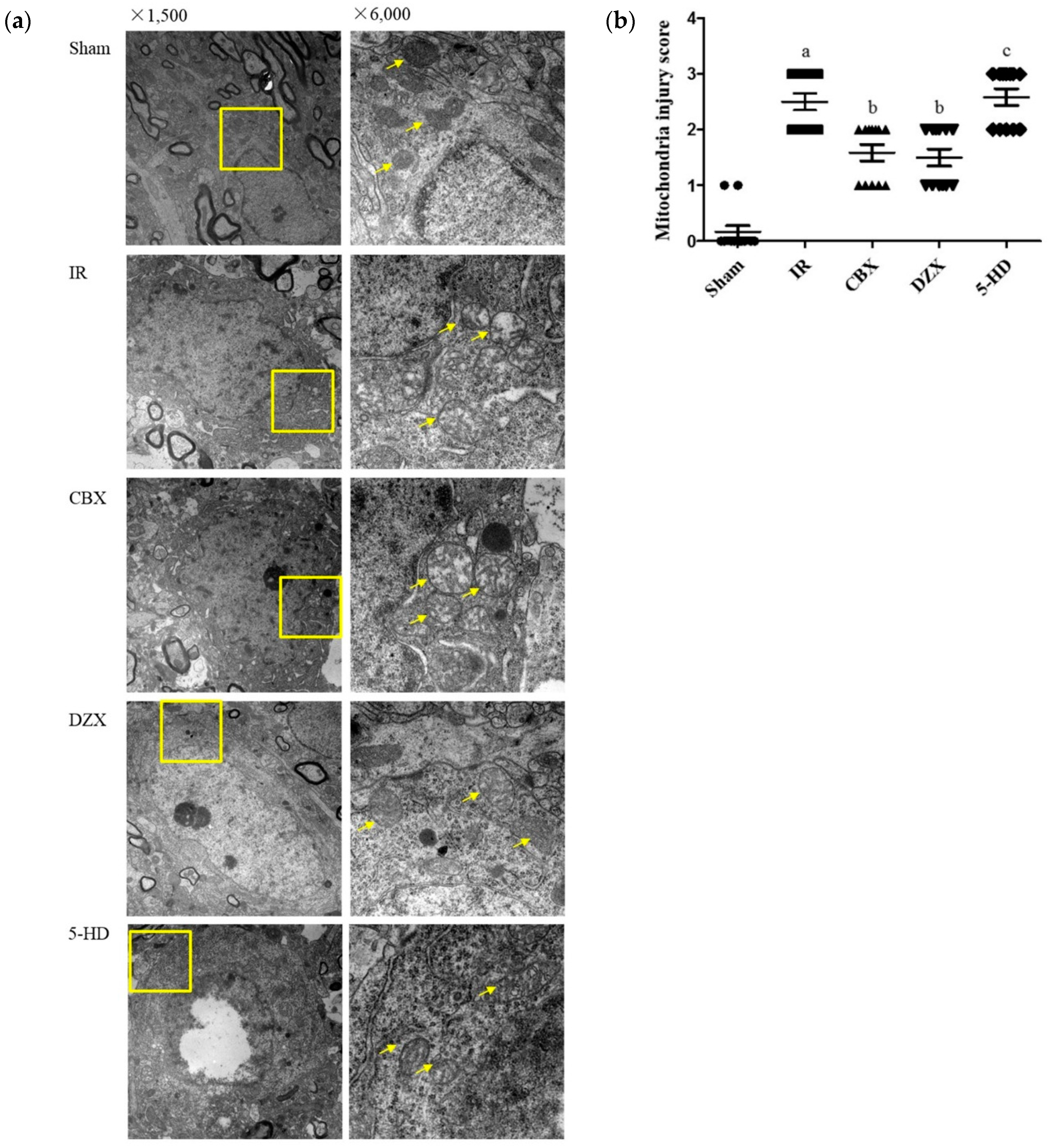
2.3. Ultrastructural Damage of the Cell Mitochondria under Transmission Electron Microscopy
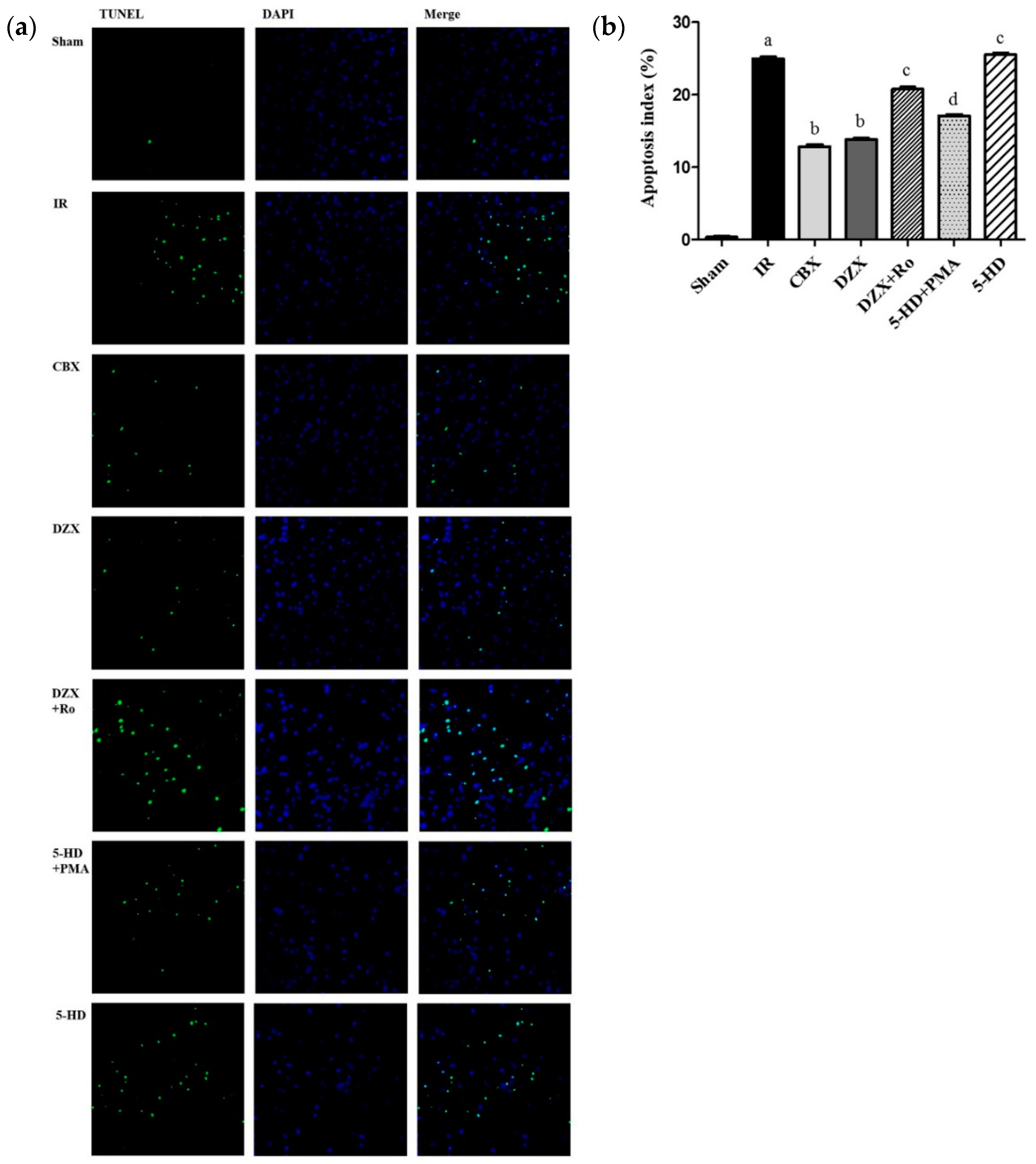
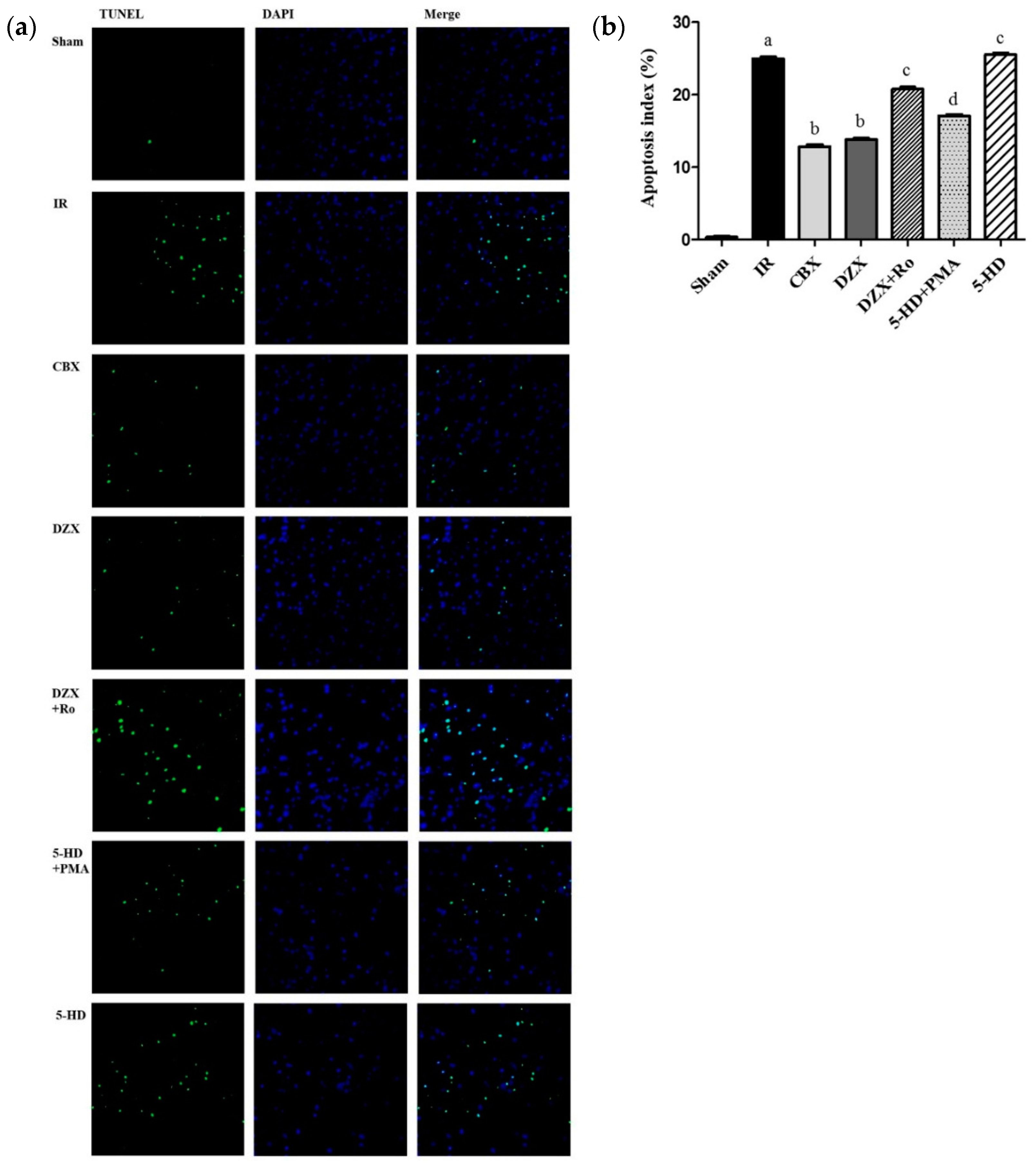
2.4. Effect of Interference on Nerovascular Unit Apoptosis Following IR Injury
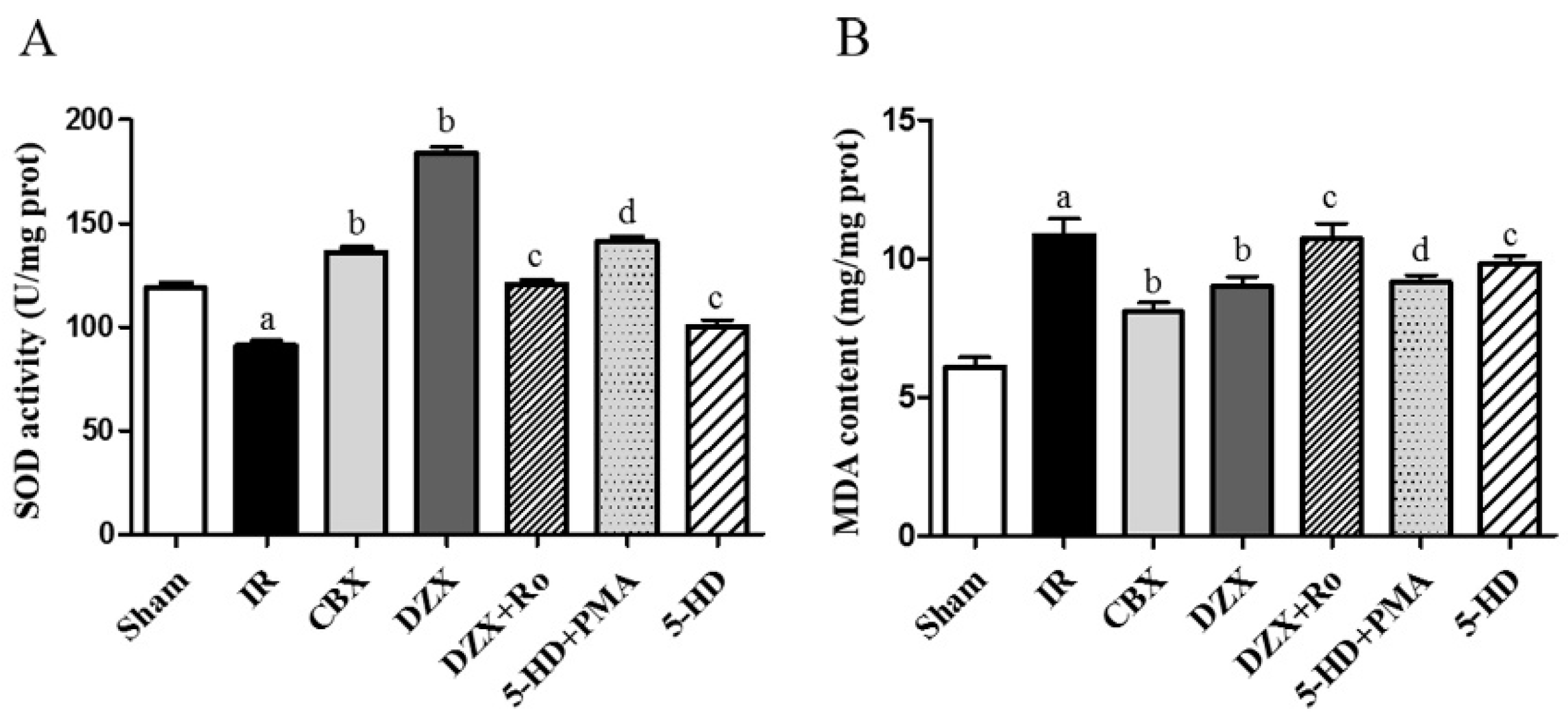
2.5. Superoxide Dismutase (SOD) Activity and Malondialdehyde (MDA) Content
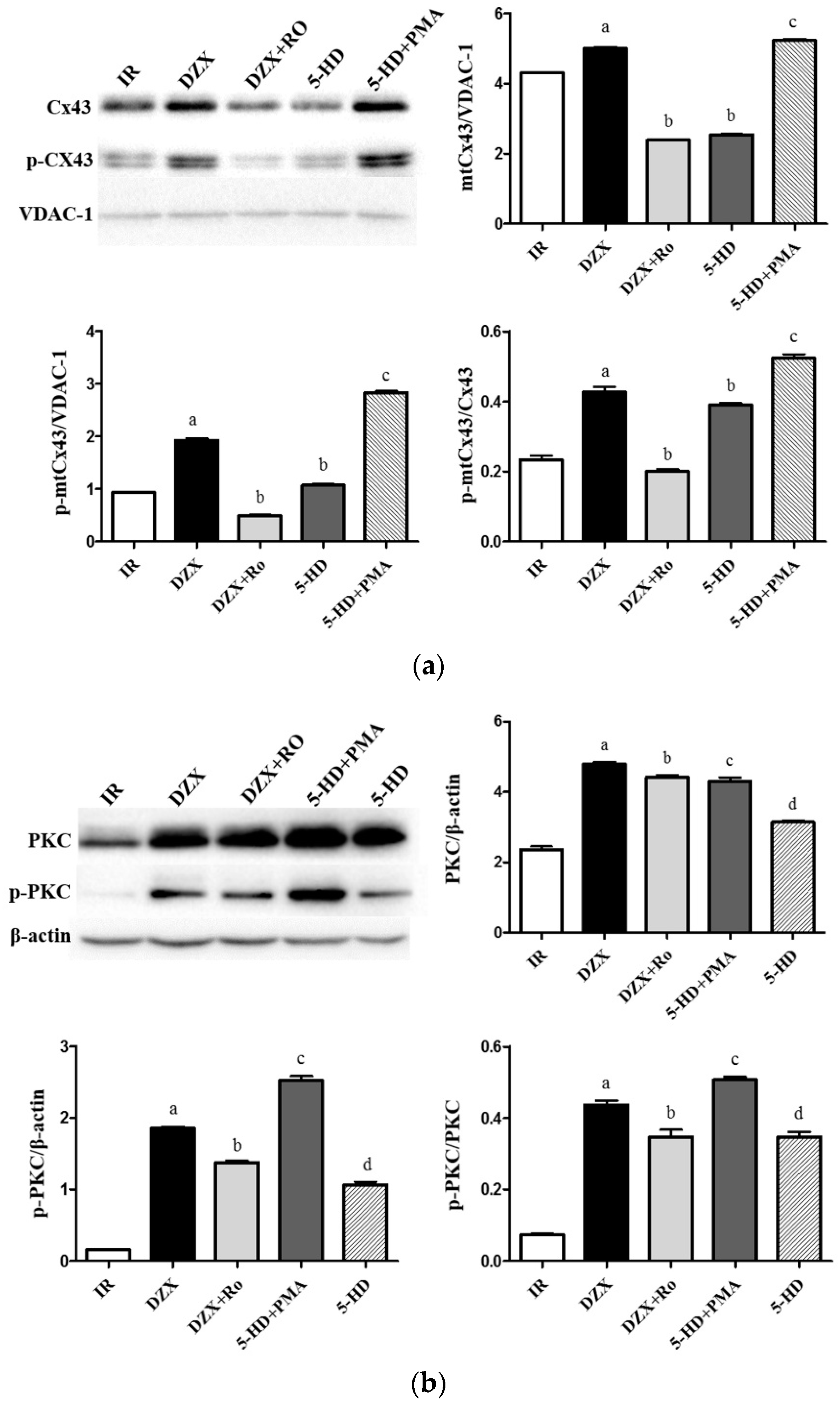
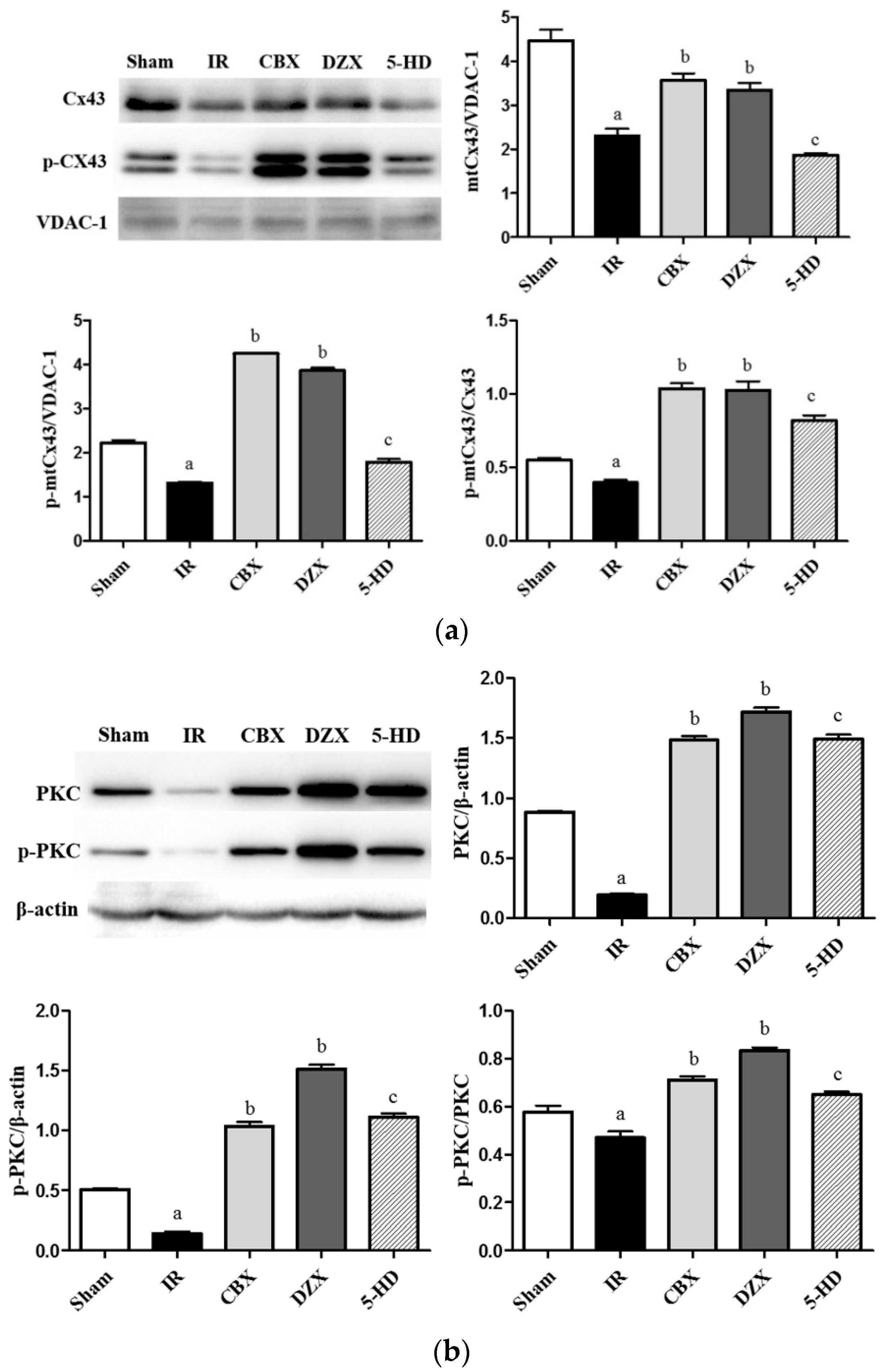
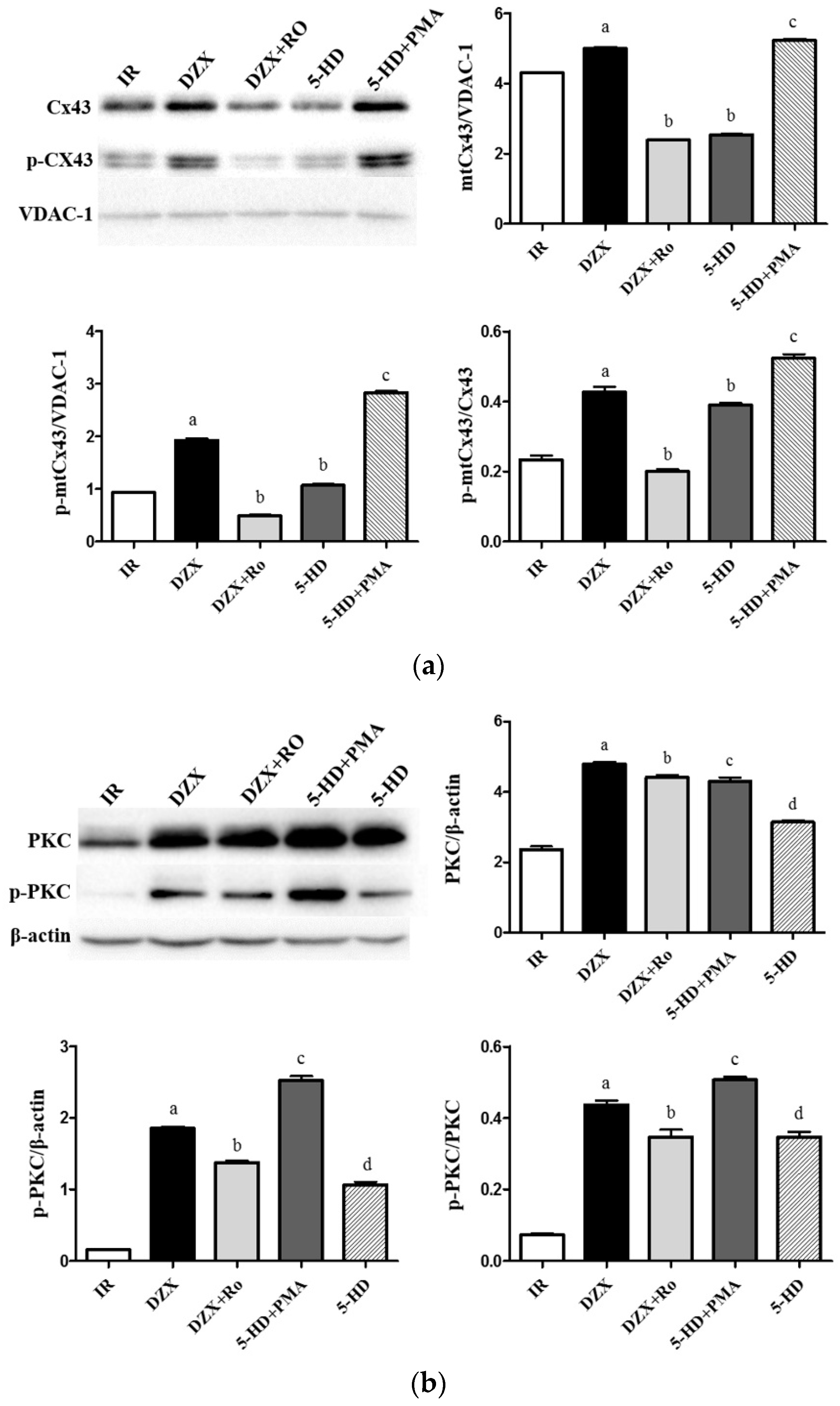
2.6. Expression of mtCx43, p-mtCx43, PKCε, and p-PKCε
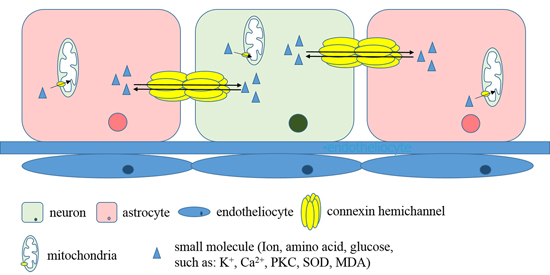
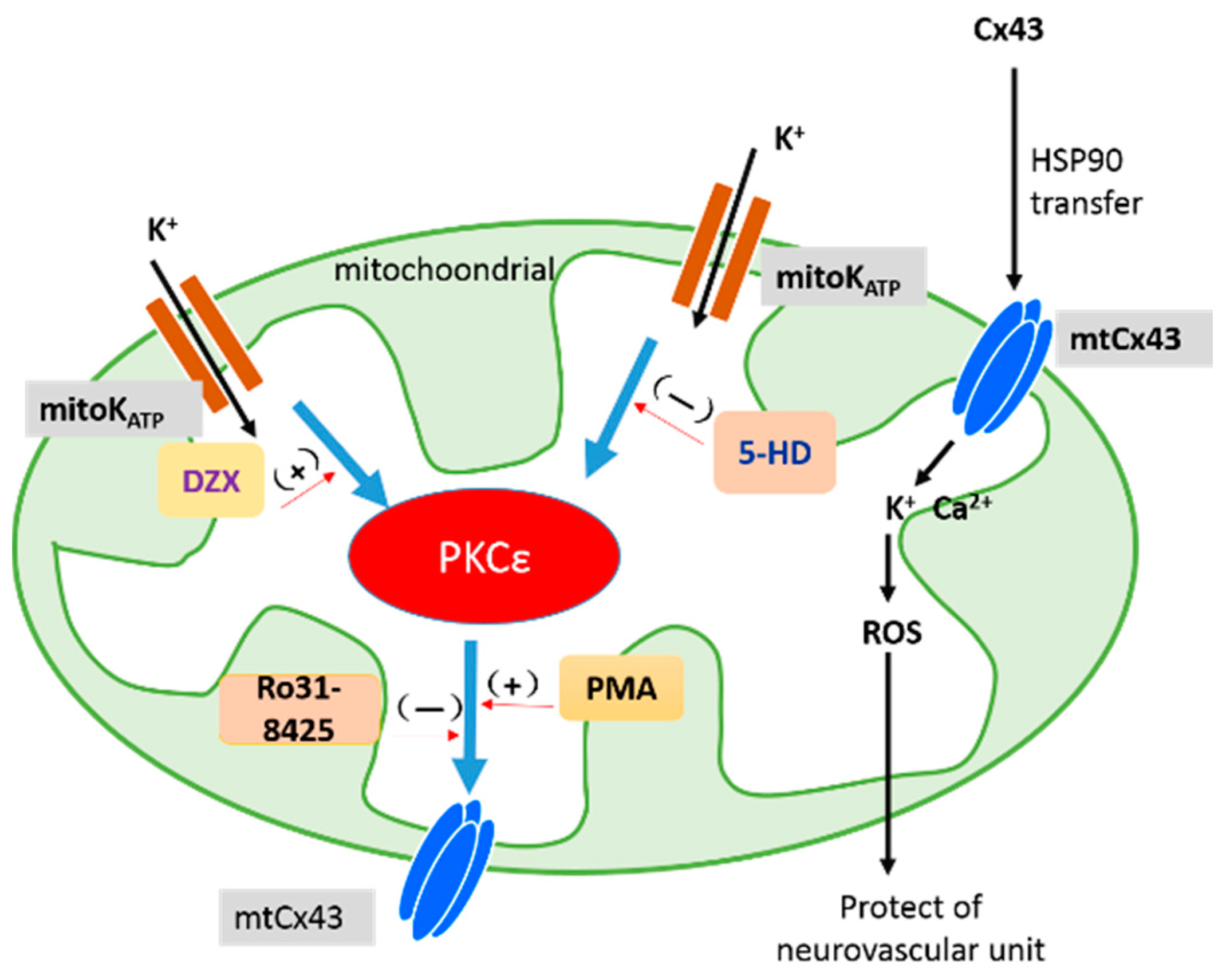
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. MCAO Model
4.4. Neurological Evaluation
4.5. TTC Staining
4.6. Mitochondria Isolation
4.7. Analysis of Mitochondrial Ultrastructure by TEM
4.8. TUNEL Staining in Cortical Neurovascular Unit Apoptosis
4.9. Detection of SOD Activity and MDA Content
4.10. Western Blot Analysis
4.11. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| I/R | ischemia-reperfusion |
| Cx43 | connexin 43 |
| mtCx43 | mitochondrial connexin 43 |
| GJ | gap junction |
| K-ATP | ATP-sensitive potassium channel |
| mitoKATP | mitochondrial ATP-sensitive potassium channel |
| sarcKATP | sarcolemmal ATP-sensitive potassium channel |
| 5-HD | 5-hydroxydecanoic acid |
| CBX | carbenoxolone |
| DZX | diazoxide |
| PMA | phorbol-12-myristate-13-acetate |
| Ro | Ro-31-8425 |
| PKA | protein kinase A |
| PKC | protein kinase C |
| MAPK | mitogen-activated protein kinase |
| MCAO | middle cerebral artery occlusion |
| AI | apoptosis index |
| DAPI | 4′,6-diamidino-2-phenylindole |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| MDA | malondialdehyde |
| TEM | transmission electron microscopy |
| TUNEL | transferase dUTP nick end labeling |
References
- Revel, J.P.; Karnovsky, M.J. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J. Cell Biol. 1967, 33, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Froger, N.; Ezan, P.; Jiang, J.X.; Bennett, M.V.; Naus, C.C.; Giaume, C.; Saez, J.C. Atp and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 2011, 118, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Tsuchida, M.; Lampe, P.D.; Murakami, M. Cardiomyocyte fgf signaling is required for cx43 phosphorylation and cardiac gap junction maintenance. Exp. Cell Res. 2013, 319, 2152–2165. [Google Scholar] [CrossRef] [PubMed]
- Kalvelyte, A.; Imbrasaite, A.; Bukauskiene, A.; Verselis, V.K.; Bukauskas, F.F. Connexins and apoptotic transformation. Biochem. Pharmacol. 2003, 66, 1661–1672. [Google Scholar] [CrossRef]
- Azarashvili, T.; Baburina, Y.; Grachev, D.; Krestinina, O.; Evtodienko, Y.; Stricker, R.; Reiser, G. Calcium-induced permeability transition in rat brain mitochondria is promoted by carbenoxolone through targeting connexin43. Am. J. Physiol. Cell Physiol. 2011, 300, C707–C720. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; Garcia-Dorado, D.; di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.S.; Haywood, G.A.; Andreka, P.; You, L.; Martin, P.E.; Evans, W.H.; Webster, K.A.; Bishopric, N.H. Reversible connexin 43 dephosphorylation during hypoxia and reoxygenation is linked to cellular atp levels. Circ. Res. 2004, 95, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Noma, A. Atp-regulated K+ channels in cardiac muscle. Nature 1983, 305, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Nagase, H.; Kishi, K.; Higuti, T. Atp-sensitive K+ channel in the mitochondrial inner membrane. Nature 1991, 352, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Tanonaka, K.; Taguchi, T.; Koshimizu, M.; Ando, T.; Morinaka, T.; Yogo, T.; Konishi, F.; Takeo, S. Role of an atp-sensitive potassium channel opener, ym934, in mitochondrial energy production in ischemic/reperfused heart. J. Pharmacol. Exp. Ther. 1999, 291, 710–716. [Google Scholar] [PubMed]
- Miura, T.; Liu, Y.; Kita, H.; Ogawa, T.; Shimamoto, K. Roles of mitochondrial atp-sensitive k channels and pkc in anti-infarct tolerance afforded by adenosine a1 receptor activation. J. Am. Coll. Cardiol. 2000, 35, 238–245. [Google Scholar] [CrossRef]
- Ozcan, C.; Holmuhamedov, E.L.; Jahangir, A.; Terzic, A. Diazoxide protects mitochondria from anoxic injury: Implications for myopreservation. J. Thorac. Cardiovasc. Surg. 2001, 121, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, C.; Bienengraeber, M.; Dzeja, P.P.; Terzic, A. Potassium channel openers protect cardiac mitochondria by attenuating oxidant stress at reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H531–H539. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, O.; Qin, Q.; Krieg, T.; Yang, X.M.; Philipp, S.; Critz, S.D.; Cohen, M.V.; Downey, J.M. Bradykinin induces mitochondrial ros generation via no, cgmp, pkg, and mitokatp channel opening and leads to cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H468–H476. [Google Scholar] [CrossRef] [PubMed]
- Rottlaender, D.; Boengler, K.; Wolny, M.; Michels, G.; Endres-Becker, J.; Motloch, L.J.; Schwaiger, A.; Buechert, A.; Schulz, R.; Heusch, G.; et al. Connexin 43 acts as a cytoprotective mediator of signal transduction by stimulating mitochondrial katp channels in mouse cardiomyocytes. J. Clin. Investig. 2010, 120, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Ahmad Waza, A.; Andrabi, K.; ul Hussain, M. Adenosine-triphosphate-sensitive K+ channel (kir6.1): A novel phosphospecific interaction partner of connexin 43 (cx43). Exp. Cell Res. 2012, 318, 2559–2566. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992, 258, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. Protein kinase c and lipid signaling for sustained cellular responses. FASEB J. 1995, 9, 484–496. [Google Scholar] [PubMed]
- Bogoyevitch, M.A.; Parker, P.J.; Sugden, P.H. Characterization of protein kinase c isotype expression in adult rat heart. Protein kinase C-epsilon is a major isotype present, and it is activated by phorbol esters, epinephrine, and endothelin. Circ. Res. 1993, 72, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Chen, H. Polydatin attenuates H2O2-induced oxidative stress via PKC pathway. Oxid. Med. Cell. Longev. 2016, 2016, 5139458. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.A.; van Veen, T.A.; de Bakker, J.M.; van Rijen, H.V. Cardiac connexins and impulse propagation. J. Mol. Cell. Cardiol. 2010, 48, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Sirnes, S.; Kjenseth, A.; Leithe, E.; Rivedal, E. Interplay between PKC and the map kinase pathway in connexin43 phosphorylation and inhibition of gap junction intercellular communication. Biochem. Biophys. Res. Commun. 2009, 382, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Wei, J.; Zhang, M.; Lin, L.; Yan, R.; Zhang, R.; Zhu, Y.H. Suppression of PKCepsilon-mediated mitochondrial connexin 43 phosphorylation at serine 368 is involved in myocardial mitochondrial dysfunction in a rat model of dilated cardiomyopathy. Mol. Med. Rep. 2015, 11, 4720–4726. [Google Scholar] [PubMed]
- Wang, Y.K.; Deng, F.; Miao, J.; Xie, H.; Feng, J.C. Neuroprotection by carbenoxolone against ischemia injury involves pi3k/Akt pathway. Clin. Lab. 2015, 61, 1561–1568. [Google Scholar] [PubMed]
- Ferdinandy, P.; Schulz, R.; Baxter, G.F. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol. Rev. 2007, 59, 418–458. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Jiao, Q.B.; Honda, T.; Kurotani, R.; Toyota, E.; Okumura, S.; Takeya, T.; Minamisawa, S.; Lanier, S.M.; Ishikawa, Y. Activator of G protein signaling 8 (ags8) is required for hypoxia-induced apoptosis of cardiomyocytes role of g beta gamma and connexin 43 (cx43). J. Biol. Chem. 2009, 284, 31431–31440. [Google Scholar] [CrossRef] [PubMed]
- Danon, A.; Zeevi-Levin, N.; Pinkovich, D.Y.; Michaeli, T.; Berkovich, A.; Flugelman, M.; Eldar, Y.C.; Rosen, M.R.; Binah, O. Hypoxia causes connexin 43 internalization in neonatal rat ventricular myocytes. Gen. Physiol. Biophys. 2010, 29, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Paucek, P.; Yarov-Yarovoy, V.; Murray, H.N.; Darbenzio, R.B.; D’Alonzo, A.J.; Lodge, N.J.; Smith, M.A.; Grover, G.J. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ. Res. 1997, 81, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sato, T.; O’Rourke, B.; Marban, E. Mitochondrial ATP-dependent potassium channels: Novel effectors of cardioprotection? Circulation 1998, 97, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; O’Rourke, B.; Marban, E. Modulation of mitochondrial ATP-dependent K+ channels by protein kinase c. Circ. Res. 1998, 83, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Z.; Feng, M.; Ren, K.; Shen, G.; Zhao, C.; Jin, X.; Jiang, K. Opening of astrocytic mitochondrial ATP-sensitive potassium channels upregulates electrical coupling between hippocampal astrocytes in rat brain slices. PLoS ONE 2013, 8, e56605. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Schalper, K.A.; Retamal, M.A.; Orellana, J.A.; Shoji, K.F.; Bennett, M.V. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp. Cell Res. 2010, 316, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Meana, M.; Rodriguez-Sinovas, A.; Cabestrero, A.; Boengler, K.; Heusch, G.; Garcia-Dorado, D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2008, 77, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Waza, A.A.; Andrabi, K.; ul Hussain, M. Protein kinase c (pkc) mediated interaction between conexin43 (cx43) and K+ (ATP) channel subunit (kir6.1) in cardiomyocyte mitochondria: Implications in cytoprotection against hypoxia induced cell apoptosis. Cell Signal. 2014, 26, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Flameng, W.; Borgers, M.; Daenen, W.; Stalpaert, G. Ultrastructural and cytochemical correlates of myocardial protection by cardiac hypothermia in man. J. Thorac. Cardiovasc. Surg. 1980, 79, 413–424. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group (n = 6) | SOD Activity (U/mg Protein) | MDA Content (mmol/mg Protein) |
|---|---|---|
| Sham | 118.9 ± 2.3 | 6.1 ± 0.3 |
| IR | 91.0 ± 2.5 a | 10.8 ± 0.6 a |
| CBX | 136.3 ± 2.2 b | 8.1 ± 0.3 b |
| DZX | 183.7 ± 3.1 b | 8.9 ± 0.3 b |
| DZX + Ro | 120.5 ± 0.2 c | 10.7 ± 0.6 c |
| 5-HD + PMA | 141.2 ± 2.3 d | 9.1 ± 0.3 d |
| 5-HD | 100.3 ± 3.1 c | 9.8 ± 0.3 c |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, S.; Shen, P.-P.; Zhao, M.-M.; Liu, X.-P.; Xie, H.-Y.; Deng, F.; Feng, J.-C. Mechanism of Mitochondrial Connexin43′s Protection of the Neurovascular Unit under Acute Cerebral Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2016, 17, 679. https://doi.org/10.3390/ijms17050679
Hou S, Shen P-P, Zhao M-M, Liu X-P, Xie H-Y, Deng F, Feng J-C. Mechanism of Mitochondrial Connexin43′s Protection of the Neurovascular Unit under Acute Cerebral Ischemia-Reperfusion Injury. International Journal of Molecular Sciences. 2016; 17(5):679. https://doi.org/10.3390/ijms17050679
Chicago/Turabian StyleHou, Shuai, Ping-Ping Shen, Ming-Ming Zhao, Xiu-Ping Liu, Hong-Yan Xie, Fang Deng, and Jia-Chun Feng. 2016. "Mechanism of Mitochondrial Connexin43′s Protection of the Neurovascular Unit under Acute Cerebral Ischemia-Reperfusion Injury" International Journal of Molecular Sciences 17, no. 5: 679. https://doi.org/10.3390/ijms17050679