
G Protein-Coupled Receptor Signaling in Stem Cells and Cancer


Abstract
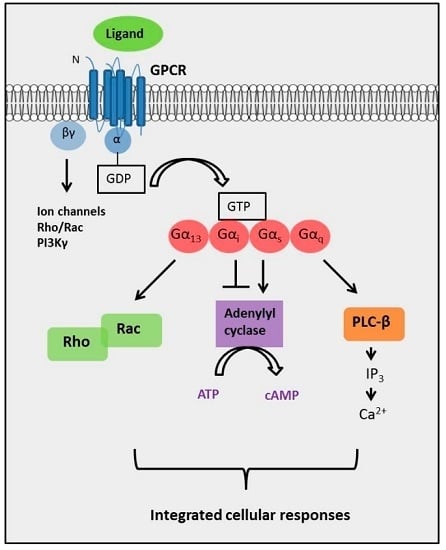
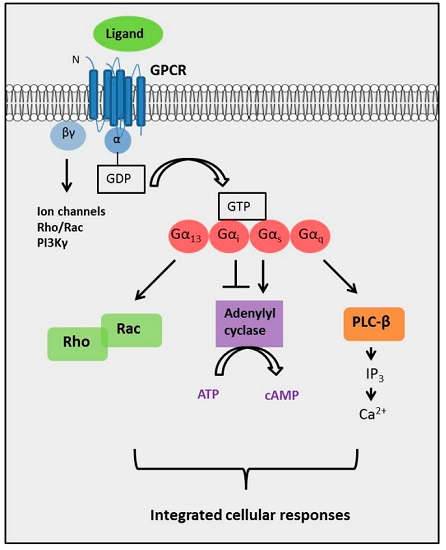
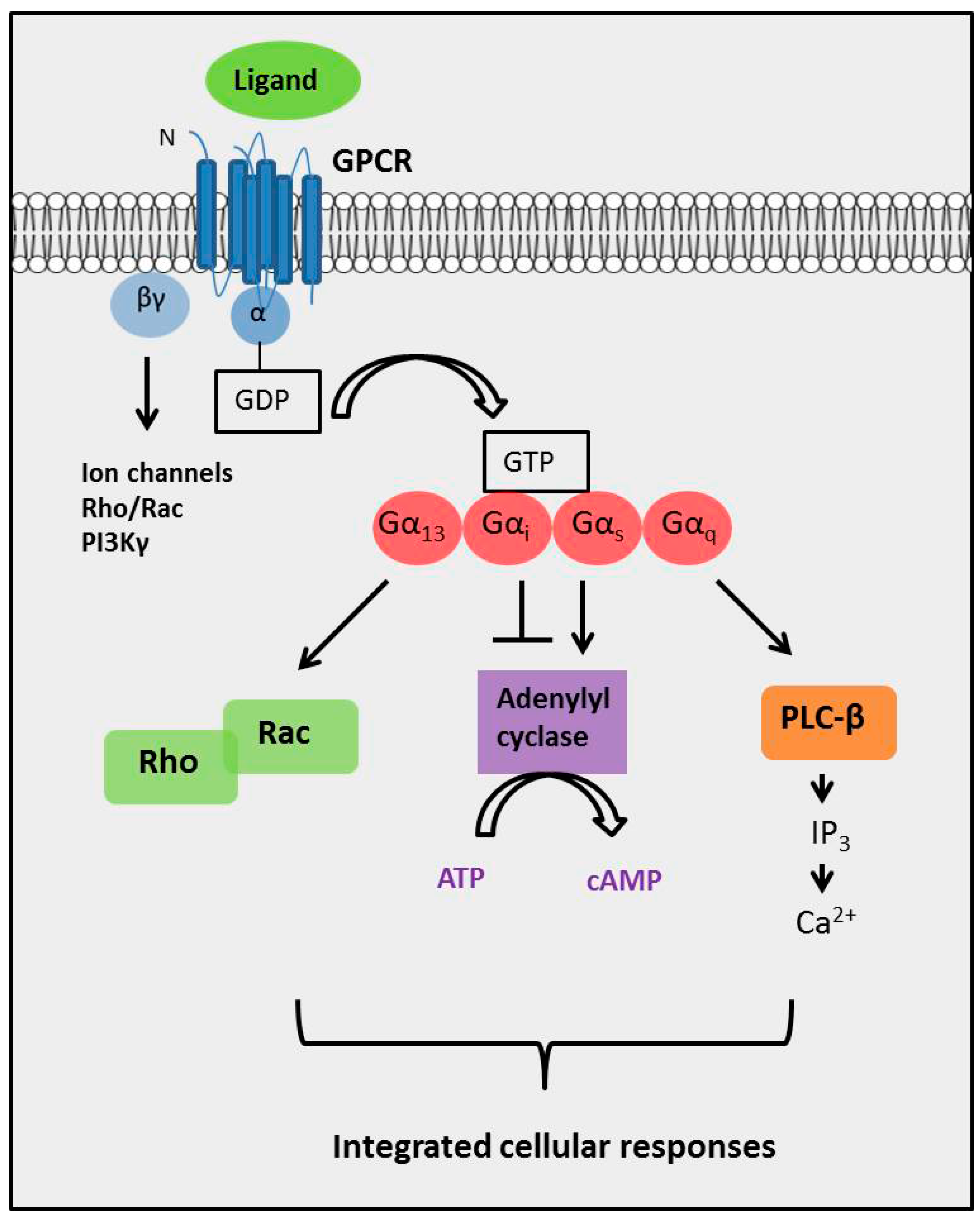
:
1. Introduction
2. The Diversity of GPCR Signaling Mechanisms
2.1. GPCR-Mediated Regulation of Stem Cell Properties
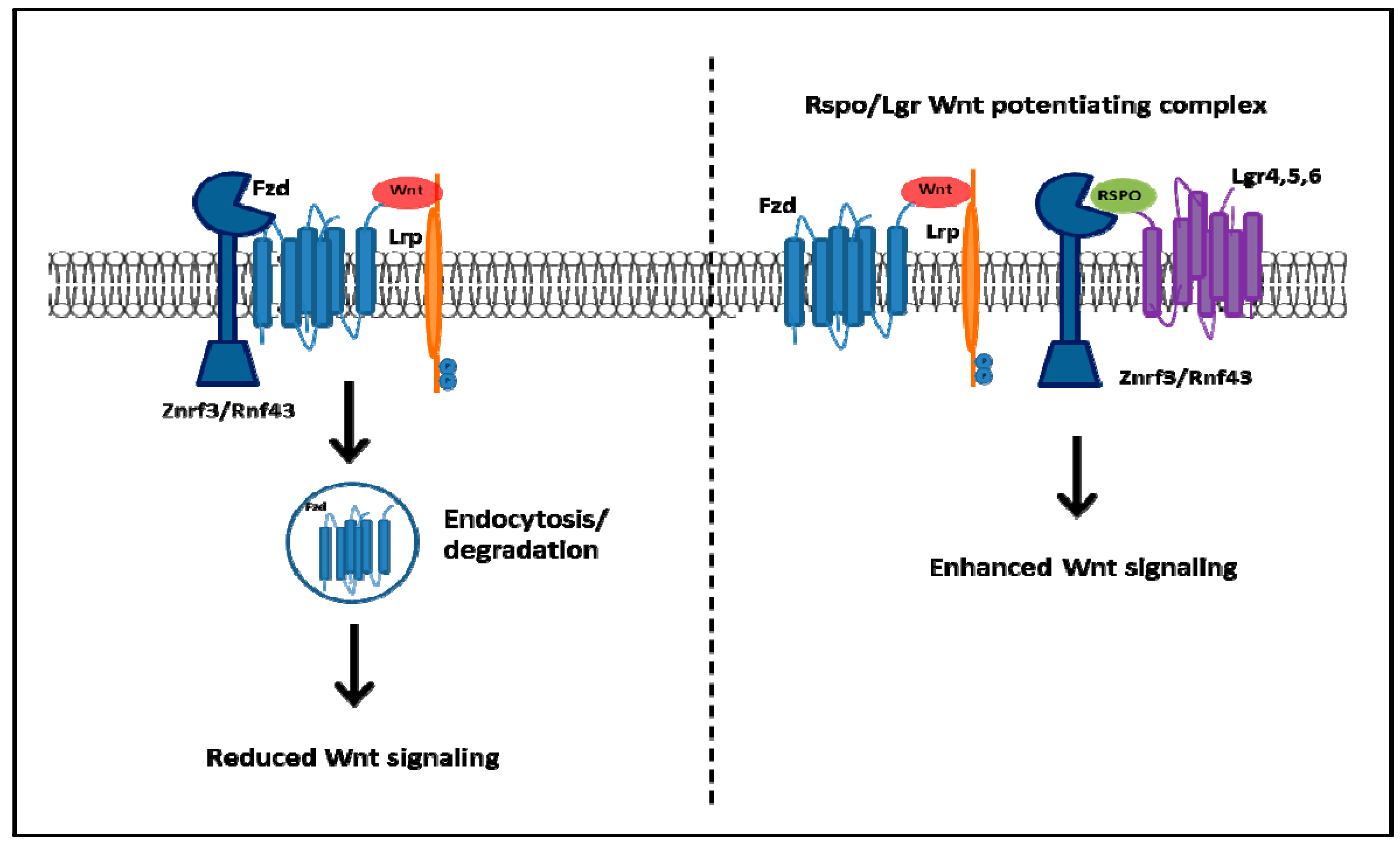
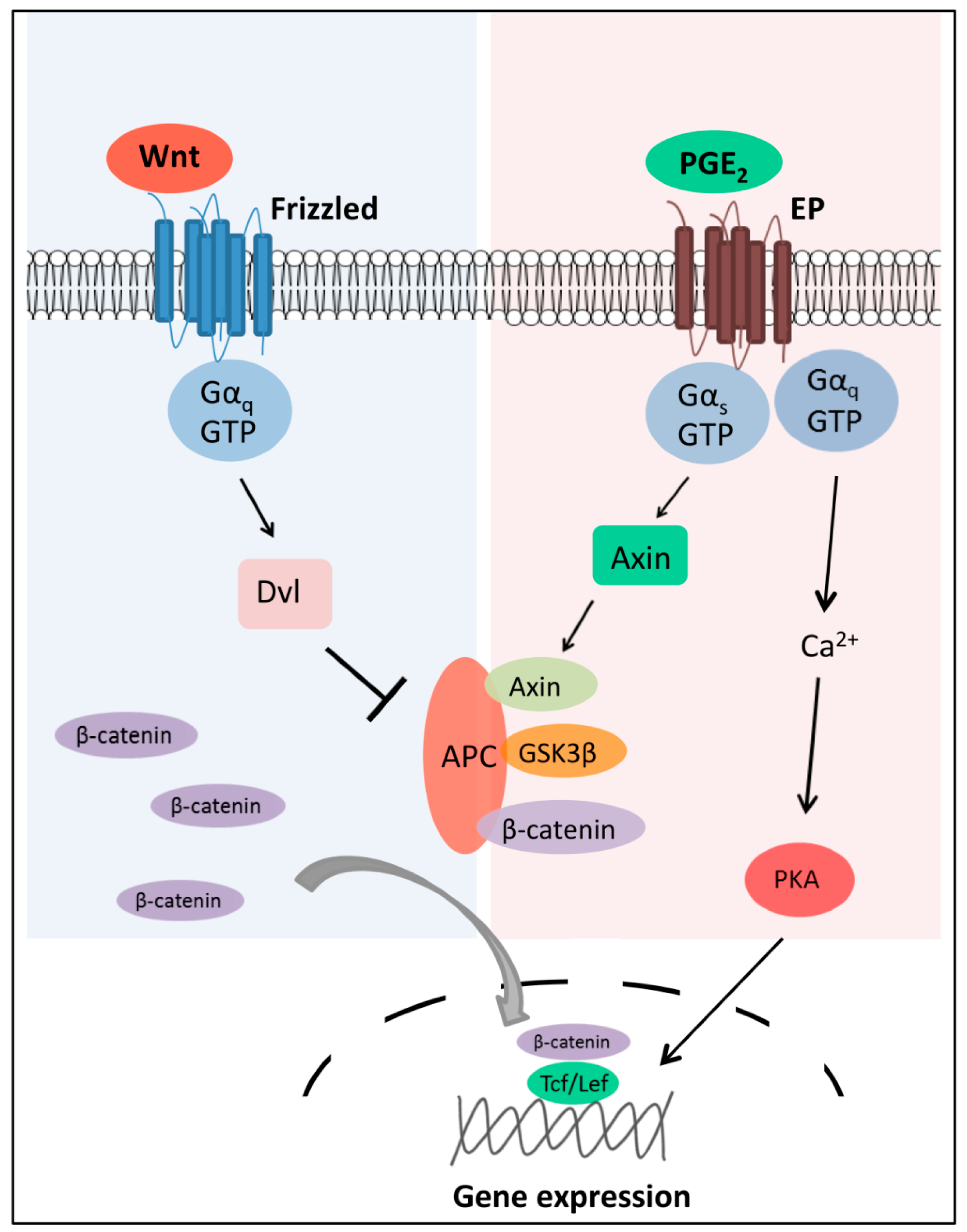
2.2. Wnt-Activated Fzd Signaling
2.3. Rhodospin Class of GPCRs
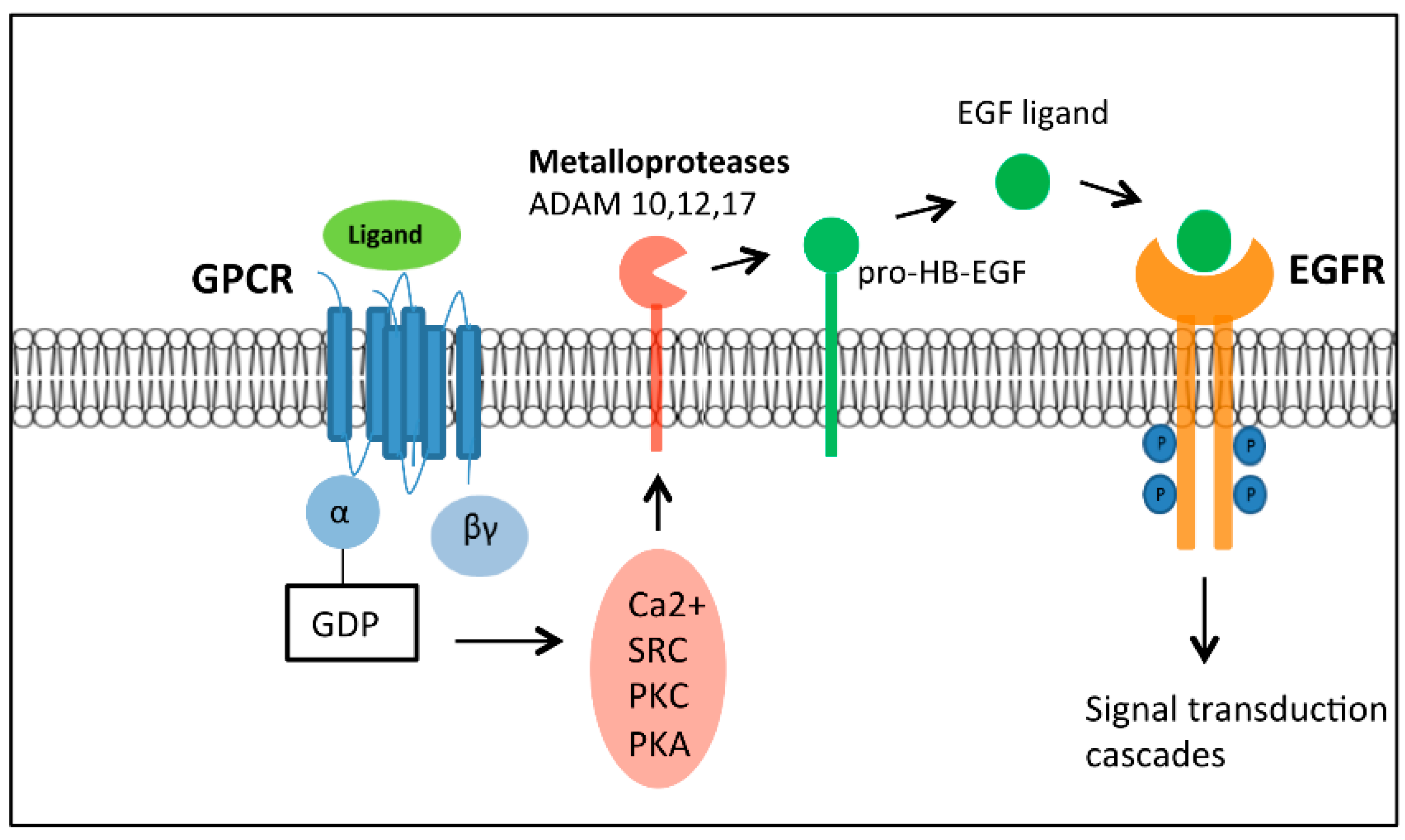
2.4. The Complexity of GPCR-Mediated Signaling
3. Clinical Implications of GPCR-Mediated Regulation of CSCs
Acknowledgments
Conflicts of Interest
References
- Wilson, A.; Trumpp, A. Bone-marrow haematopoietic-stem-cell niches. Nat. Rev. Immunol. 2006, 6, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Reubinoff, B.E.; Pera, M.F.; Fong, C.Y.; Trounson, A.; Bongso, A. Embryonic stem cell lines from human blastocysts: Somatic differentiation in vitro. Nat. Biotechnol. 2000, 18, 399–404. [Google Scholar] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Jahagirdar, B.N.; Reinhardt, R.L.; Schwartz, R.E.; Keene, C.D.; Ortiz-Gonzalez, X.R.; Reyes, M.; Lenvik, T.; Lund, T.; Blackstad, M.; et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002, 418, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Blau, H.M.; Brazelton, T.R.; Weimann, J.M. The evolving concept of a stem cell: Entity or function? Cell 2001, 105, 829–841. [Google Scholar] [CrossRef]
- Verfaillie, C.M.; Pera, M.F.; Lansdorp, P.M. Stem cells: Hype and reality. ASH Educ. Program Book 2002, 1, 369–391. [Google Scholar] [CrossRef]
- Jensen, U.B.; Lowell, S.; Watt, F.M. The spatial relationship between stem cells and their progeny in the basal layer of human epidermis: A new view based on whole-mount labelling and lineage analysis. Development 1999, 126, 2409–2418. [Google Scholar] [PubMed]
- Watt, F.M.; Hogan, B.L.M. Out of Eden: Stem cells and their niches. Science 2000, 287, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Salomonis, N.; Tomoda, K.; Yamanaka, S.; Conklin, B.R. G(I)-coupled GPCR signaling controls the formation and organization of human pluripotent colonies. PLoS ONE 2009, 4, e7780. [Google Scholar] [CrossRef] [PubMed]
- Layden, B.T.; Newman, M.; Chen, F.; Fisher, A.; Lowe, W.L., Jr. G protein coupled receptors in embryonic stem cells: A role for Gs-α signaling. PLoS ONE 2010, 5, e9105. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulou, T.; Priestley, G.V.; Bonig, H.; Nakamoto, B. The role of G-protein signaling in hematopoietic stem/progenitor cell mobilization. Blood 2003, 101, 4739–4747. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, S.; Peng, S.B. Overexpression of G protein-coupled receptors in cancer cells: Involvement in tumor progression. Int. J. Oncol. 2005, 27, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Dirks, P.B. Cancer: Stem cells and brain tumours. Nature 2006, 444, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Domen, J.; Gandy, K.L.; Weissman, I.L. Systemic overexpression of BCL-2 in the hematopoietic system protects transgenic mice from the consequences of lethal irradiation. Blood 1998, 91, 2272–2282. [Google Scholar] [PubMed]
- Domen, J.; Weissman, I.L. Hematopoietic stem cells need two signals to prevent apoptosis; BCL-2 can provide one of these, Kitl/c-Kit signaling the other. J. Exp. Med. 2000, 192, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Caceres-Cortes, J.; Mindeni, M.; Patersoni, B.; Caligiuri, M.A. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.I.; Strathmann, M.P.; Gautam, N. Diversity of G-proteins in signal transduction. Science 1991, 252, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Taussig, R.; Iniguez-Lluhi, J.A.; Gilman, A.G. Inhibition of adenylyl cyclase by Gi α. Science 1993, 261, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Hamm, H.E. The many faces of G protein signaling. J. Biol. Chem. 1998, 273, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Kozasa, T.; Jiang, X.; Hart, M.J.; Strenweis, P.M.; Singer, W.D.; Gilman, A.G.; Bollag, G.; Sternweis, P.C. p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13. Science 1998, 280, 2109–2111. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Vera, T.M.; Vanhauwe, J.; Thomas, T.O.; Medkova, M.; Preininger, A.; Mazzoni, M.R.; Hamm, H.E. Insights into G protein structure, function, and regulation. Endocr. Rev. 2003, 24, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Poised to signal. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E395–E402. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.K. Cross-talk between calcium and protein kinase A in the regulation of cell migration. Curr. Opin. Cell Biol. 2011, 23, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Strasser, R.H.; Benovic, J.L.; Caron, M.G.; Lefkowitz, R.J. β-agonist- and prostaglandin E1-induced translocation of the β-adrenergic receptor kinase: Evidence that the kinase may act on multiple adenylate cyclase-coupled receptors. Proc. Natl. Acad. Sci. USA 1986, 83, 6362–6366. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L.; Strasser, R.H.; Caron, M.G.; Lefkowitz, R.J. B-adrenergic receptor kinase: Identification of a novel protein kinase that phosphorylates the agonist-occupied form of the receptor. Proc. Natl. Acad. Sci. USA 1986, 83, 2797–2801. [Google Scholar] [CrossRef] [PubMed]
- Attramadal, H.; Arriza, J.L.; Aoki, C.; Dawson, T.M.; Codina, J.; Kwatra, M.M.; Snyder, S.H.; Caron, M.G.; Lefkowitz, R.J. B-arrestin2, a novel member of the arrestin/β-arrestin gene family. J. Biol. Chem. 1992, 267, 17882–17890. [Google Scholar] [PubMed]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. B-Arrestin: A protein that regulates β-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Posner, B.A.; Mukhopadhyay, S.; Tesmer, J.J.; Gilman, A.G.; Ross, E.M. Modulation of the affinity and selectivity of RGS protein interaction with Gα subunits by a conserved asparagine/serine residue. Biochemistry 1999, 38, 7773–7779. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.A.; Baragli, A.; Bernstein, L.S.; Hepler, J.R.; Hébert, T.E.; Chidiac, P. RGS2 interacts with Gs and adenylyl cyclase in living cells. Cell Signal. 2006, 18, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Schoeber, J.P.; Topala, C.N.; Wang, X.; Diepens, R.J.; Lambers, T.T.; Hoenderop, J.G.; Bindels, R.J. RGS2 inhibits the epithelial Ca2+ channel TRPV6. J. Biol. Chem. 2006, 281, 29669–29674. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Wade, S.M.; Woolf, P.J.; Linderman, J.J.; Traynor, J.R.; Neubig, R.R. A spatial focusing model for G protein signals. Regulator of G protein signaling (RGS) protien-mediated kinetic scaffolding. J. Biol. Chem. 2003, 278, 7278–7284. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Herr, D.; Mutoh, T.; Chun, J. Lysophosphatidic acid (LPA) and its receptors. Curr. Opin. Pharmacol. 2009, 9, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, T.; Hla, T. Structural and functional characteristics of S1P receptors. J. Cell. Biochem. 2004, 92, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.; Thomas, S.M.; Zhang, Q.; Wentzel, A.L.; Siegfried, J.M.; Li, J.Y.; Grandis, J.R. Mitogenic effects of gastrin-releasing peptide in head and neck squamous cancer cells are mediated by activation of the epidermal growth factor receptor. Oncogene 2003, 22, 6183–6193. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Kitayama, J.; Yamaguchi, H.; Okaji, Y.; Tsuno, N.H.; Watanabe, T.; Takuwa, Y.; Nagawa, H. Lysophosphatidic acid (LPA) enhances the metastatic potential of human colon carcinoma DLD1 cells through LPA1. Cancer Res. 2003, 63, 1706–1711. [Google Scholar] [PubMed]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.A.; Coleman, D.E.; Lee, E.; Iñiguez-Lluhi, J.A.; Posner, B.A.; Gilman, A.G.; Sprang, S.R. The structure of the G protein heterotrimer G iα1 β 1γ2. Cell 1995, 83, 1047–1058. [Google Scholar] [CrossRef]
- Lambright, D.G.; Sondek, J.; Bohm, A.; Skiba, N.P.; Hamm, H.E.; Sigler, P.B. The 2.0 Å crystal structure of a heterotrimeric G protein. Nature 1996, 379, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Kjeldgaard, M.; Nyborg, J.; Clark, B.F. The GTP binding motif: Variations on a theme. FASEB J. 1996, 10, 1347–1368. [Google Scholar] [PubMed]
- Harhammer, R.; Gohla, A.; Schultz, G. Interaction of G protein G β γ dimers with small GTP-binding proteins of the Rho family. FEBS Lett. 1996, 399, 211–214. [Google Scholar] [CrossRef]
- Katoh, M. WNT signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Mitalipova, M.; Noggle, S.; Tibbitts, D.; Venable, A.; Rao, R.; Stice, S.L. Long-term proliferation of human embryonic stem cell-derived neuroepithelial cells using defined adherent culture conditions. Stem Cells 2006, 24, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Dhara, S.K.; Stice, S.L. Neural differentiation of human embryonic stem cells. J. Cell. Biochem. 2008, 105, 633–640. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.D.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Liu, T.; Malbon, C.C. Structure-function analysis of Frizzleds. Cell Signal. 2006, 18, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.T.; Anastas, J.N.; Toroni, R.A.; Shinohara, M.M.; Goodson, J.M.; Bosserhoff, A.K.; Chien, A.J.; Moon, R.T. WLS inhibits melanoma cell proliferation through the β-catenin signalling pathway and induces spontaneous metastasis. EMBO Mol. Med. 2012, 4, 1294–1307. [Google Scholar] [CrossRef] [PubMed]
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-β-catenin signaling axis. Science 2005, 310, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Katanaev, V.L.; Ponzielli, R.; Semeriva, M.; Tomlinson, A. Trimeric G protein-dependent frizzled signaling in Drosophila. Cell 2005, 120, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Foord, S.M.; Bonner, T.I.; Neubig, R.R.; Rosser, E.M.; Pin, J.P.; Davenport, A.P.; Spedding, M.; Harmar, A.J. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol. Rev. 2005, 57, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Nusse, R. Wnt Proteins. Cold Spring Harb. Perspect. Biol. 2012, 4, a007864. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Yamamoto, H.; Kishida, S. Multiplicity of the interactions of Wnt proteins and their receptors. Cell Signal. 2007, 19, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, P.M.; Williams, J.; Xu, Q.; Leahy, D.J.; Nathans, J. Mutational analysis of Norrin-Frizzled4 recognition. J. Biol. Chem. 2007, 282, 4057–4068. [Google Scholar] [CrossRef] [PubMed]
- Slusarski, D.C.; Corces, V.G.; Moon, R.T. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature 1997, 390, 410–413. [Google Scholar] [PubMed]
- Kohn, A.D.; Moon, R.T. Wnt and calcium signaling: β-catenin-independent pathways. Cell Calcium 2005, 38, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Seifert, J.R.; Mlodzik, M. Frizzled/PCP signalling: A conserved mechanism regulating cell polarity and directed motility. Nat. Rev. Genet. 2007, 8, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Kemp, C.R.; Willems, E.; Wawrzak, D.; Hendrickx, M.; Agbor Agbor, T.; Leyns, L. Expression of Frizzled5, Frizzled7, and Frizzled10 during early mouse development and interactions with canonical Wnt signaling. Dev. Dyn. 2007, 236, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huso, D.; Cahill, H.; Ryugo, D.; Nathans, J. Progressive cerebellar, auditory, and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J. Neurosci. 2001, 21, 4761–4771. [Google Scholar] [PubMed]
- Ranheim, E.A.; Kwan, H.C.; Reya, T.; Wang, Y.K.; Weissman, I.L.; Francke, U. Frizzled 9 knock-out mice have abnormal B-cell development. Blood 2005, 105, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Nathans, J. An essential role for frizzled 5 in mammalian ocular development. Development 2008, 135, 3567–3576. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Meijer, L.; Skaltsounis, L.; Greengard, P.; Brivanlou, A.H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 2004, 10, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Melchior, K.; Weiss, J.; Zaehres, H.; Kim, Y.M.; Lutzko, C.; Roosta, N.; Hescheler, J.; Muschen, M. The WNT receptor FZD7 contributes to self-renewal signaling of human embryonic stem cells. Biol. Chem. 2008, 389, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Goss, A.M.; Cohen, E.D.; Kadzik, R.; Lepore, J.J.; Muthukumaraswamy, K.; Yang, J.; de Mayo, F.J.; Whitsett, J.A.; Parmacek, M.S.; et al. A Gata6-Wnt pathway required for epithelial stem cell development and airway regeneration. Nat. Genet. 2008, 40, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Borello, U.; Berarducci, B.; Murphy, P.; Bajard, L.; Buffa, V.; Piccolo, S.; Buckingham, M.; Cossu, G. The Wnt/β-catenin pathway regulates Gli-mediated Myf5 expression during somitogenesis. Development 2006, 133, 3723–3732. [Google Scholar] [CrossRef] [PubMed]
- Vijayaragavan, K.; Szabo, E.; Bosse, M.; Ramos-Mejia, V.; Moon, R.T.; Bhatia, M. Noncanonical Wnt signaling orchestrates early developmental events toward hematopoietic cell fate from human embryonic stem cells. Cell Stem Cell 2009, 4, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.L.; Chien, A.J.; Moon, R.T. Wnt/Fz signaling and the cytoskeleton: Potential roles in tumorigenesis. Cell Res. 2009, 19, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta Rev. Cancer 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Brennan, K.R.; Brown, A.M.C. Wnt proteins in mammary development and cancer. J. Mammary Gland. Biol. Neoplas. 2004, 9, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/β-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Rodriguez, M.; Valdez, J.M.; Zhu, X.; Tan, K.; Li, D.; Siwko, S.; Xin, L.; Liu, M. Lgr4 is a key regulator of prostate development and prostate stem cell differentiation. Stem Cells 2013, 31, 2492–2505. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Fuerer, C.; Ching, W.; Harnish, K.; Logan, C.; Zeng, A.; Ten Berge, D.; Kalani, Y. Wnt signaling and stem cell control. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Eaves, C.J.; Humphries, R.K. Acute myeloid leukemia and the Wnt pathway. N. Engl. J. Med. 2010, 362, 2326–2327. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.A.; Yang, C.; Leung, H.H.; Lynch, J.R.; Gonzales, E.; Liu, B.; Haber, M.; Norris, M.D.; Wang, J.; Wang, J.Y. GPR84 sustains aberrant β-catenin signaling in leukemic stem cells for maintenance of MLL leukemogenesis. Blood 2014, 124, 3284–3294. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; Yi, H.; Casolari, D.A.; Voli, F.; Gonzales-Aloy, E.; Fung, T.K.; Liu, B.; Brown, A.; Liu, T.; Haber, M.; et al. GAQ signaling is required for the maintenance of MLL-AF9 induced AML. Leukemia 2016. [Google Scholar] [CrossRef] [PubMed]
- Gloriam, D.E.; Fredriksson, R.; Schioth, H.B. The G protein-coupled receptor subset of the rat genome. BMC Genom. 2007, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Jaks, V.; Barker, N.; Kasper, M.; van Es, J.H.; Snippert, H.J.; Clevers, H.; Toftgard, R. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet. 2008, 40, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Rookmaaker, M.B.; Kujala, P.; Ng, A.; Leushacke, M.; Snippert, H.; van de Wetering, M.; Tan, S.; van Es, J.H.; Huch, M.; et al. Lgr5+ ve stem/progenitor cells contribute to nephron formation during kidney development. Cell Rep. 2012, 2, 540–552. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Ciampricotti, M.; Michalak, E.M.; Tan, D.W.; Speksnijder, E.N.; Hau, C.S.; Clevers, H.; Barker, N.; Jonkers, J. Developmental stage-specific contribution of LGR5+ cells to basal and luminal epithelial lineages in the postnatal mammary gland. J. Pathol. 2012, 228, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Brenot, A.; Lawson, D.A.; Linnemann, J.R.; van Kappel, E.C.; Wong, K.C.; de Sauvage, F.; Klein, O.D.; Werb, Z. Lgr5-expressing cells are sufficient and necessary for postnatal mammary gland organogenesis. Cell Rep. 2013, 3, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.K.; Li, Y.; Redding, K.M.; Iwatsuki, K.; Margolskee, R.F.; Jiang, P. Lgr5-EGFP marks taste bud stem/progenitor cells in posterior tongue. Stem Cells 2013, 31, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Fevr, T.; Robine, S.; Louvard, D.; Huelsken, J. Wnt/β-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol. Cell. Biol. 2007, 27, 7551–7559. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; Van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [PubMed]
- Kim, K.A.; Wagle, M.; Tran, K.; Zhan, X.; Dixon, M.A.; Liu, S.; Gros, D.; Korver, W.; Yonkovich, S.; Tomasevic, N.; et al. R-Spondin family members regulate the Wnt pathway by a common mechanism. Mol. Biol. Cell. 2008, 19, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.; Zhang, H.H.; Odorizzi, A.; Burgess, A.W. LGR5 Is a Negative regulator of tumourigenicity, antagonizes Wnt signalling and regulates cell adhesion in colorectal cancer cell lines. PLoS ONE 2011, 6, e22733. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Lin, Q.S.; Thomas, A.; Liu, Q.Y. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [PubMed]
- Hull, M.A.; Ko, S.C.; Hawcroft, G. Prostaglandin EP receptors: Targets for treatment and prevention of colorectal cancer? Mol. Cancer Ther. 2004, 3, 1031–1039. [Google Scholar] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Graham, T.A.; Elia, G.; Wright, N.A.; Rodriguez-Justo, M. Characterization of LGR5 stem cells in colorectal adenomas and carcinomas. Sci. Rep. 2015, 5, 8654. [Google Scholar] [CrossRef] [PubMed]
- Kozar, S.; Morrissey, E.; Nicholson, A.M.; van der Heijden, M.; Zecchini, H.I.; Kemp, R.; Tavare, S.; Vermeulen, L.; Winton, D.J. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell 2013, 13, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Cereser, B.; Melton, S.; Fletcher, A.G.; Rodriguez-Justo, M.; Tadrous, P.J.; Humphries, A.; Elia, G.; McDonald, S.A.; Wright, N.A.; et al. Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 2014, 8, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; Haegebarth, A.; Kasper, M.; Jaks, V.; van Es, J.H.; Barker, N.; van de Wetering, M.; van den Born, M.; Begthel, H.; Vries, R.G.; et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 2010, 327, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Mazerbourg, S.; Bouley, D.M.; Sudo, S.; Klein, C.A.; Zhang, J.V.; Kawamura, K.; Goodrich, L.V.; Rayburn, H.; Tessier-Lavigne, M.; Hsueh, A.J.W. Leucine-rich repeat-containing, G protein-coupled receptor 4 null mice exhibit intrauterine growth retardation associated with embryonic and perinatal lethality. Mol. Endocrinol. 2004, 18, 2241–2254. [Google Scholar] [CrossRef] [PubMed]
- Mendive, F.; Laurent, P.; Van Schoore, G.; Skarnes, W.; Pochet, R.; Vassart, G. Defective postnatal development of the male reproductive tract in LGR4 knockout mice. Dev. Biol. 2006, 290, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Matsubara, M.; Matsuo, T.; Mohri, Y.; Kazama, I.; Hatano, R.; Umezawa, A.; Nishimori, K. Leucine-rich repeat-containing G protein-coupled receptor-4 (LGR4, Gpr48) is essential for renal development in mice. Nephron Exp. Nephrol. 2006, 104, E63–E75. [Google Scholar] [CrossRef] [PubMed]
- Mohri, Y.; Oyama, K.; Akamatsu, A.; Kato, S.; Nishimori, K. Lgr4-deficient mice showed premature differentiation of ureteric bud with reduced expression of Wnt effector Lef1 and Gata3. Dev. Dyn. 2011, 240, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.S.; Luo, J.; Cheng, X.H.; Jin, C.; Zhou, X.T.; Qu, J.; Tu, L.; Ai, D.; Li, D.L.; Wang, J.; et al. Deletion of G protein-coupled receptor 48 leads to ocular anterior segment dysgenesis (ASD) through down-regulation of Pitx2. Proc. Natl. Acad. Sci. USA 2008, 105, 6081–6086. [Google Scholar] [CrossRef] [PubMed]
- Oyama, K.; Mohri, Y.; Sone, M.; Nawa, A.; Nishimori, K. Conditional Knockout of Lgr4 Leads to Impaired Ductal Elongation and Branching Morphogenesis in Mouse Mammary Glands. Sex. Dev. 2011, 5, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xie, N.; Xie, K.; Zeng, J.; Cheng, L.; Lei, Y.; Liu, Y.; Song, L.; Dong, D.; Chen, Y.; et al. GPR48, a poor prognostic factor, promotes tumor metastasis and activates β-catenin/TCF signaling in colorectal cancer. Carcinogenesis 2013, 34, 2861–2869. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.Y.; Wang, J.L.; Kavallaris, M.; Wang, J.Y. Lgr4-mediated potentiation of Wnt/β-Catenin signaling promotes MLL leukemogenesis via an Rspo3/Wnt3a-GNAQ pathway in leukemic stem cells. Blood 2013, 122, 887. [Google Scholar]
- Lepley, D.; Paik, J.H.; Hla, T.; Ferrer, F. The G protein-coupled receptor S1P2 regulates Rho/Rho kinase pathway to inhibit tumor cell migration. Cancer Res. 2005, 65, 3788–3795. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.Y.; Jung, C.Y.; Liu, C.M.; Sheng, H.M. Prostaglandin E-2 stimulates the β-catenin/T cell factor-dependent transcription in colon cancer. J. Biol. Chem. 2005, 280, 26565–26572. [Google Scholar] [CrossRef] [PubMed]
- Nambotin, S.B.; Lefrancois, L.; Sainsily, X.; Berthillon, P.; Kim, M.; Wands, J.R.; Chevallier, M.; Jalinot, P.; Scoazec, J.Y.; Trepo, C.; et al. Pharmacological inhibition of Frizzled-7 displays anti-tumor properties in hepatocellular carcinoma. J. Hepatol. 2011, 54, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Schulte, G.; Bryja, V. The Frizzled family of unconventional G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.; Ma, X.; Kochel, T.; Goloubeva, O.; Staats, P.; Thompson, K.; Martin, S.; Reader, J.; Take, Y.; Collin, P.; et al. Prostaglandin E receptor EP4 is a therapeutic target in breast cancer cells with stem-like properties. Breast Cancer Res. Treat. 2014, 143, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Arun, C.; London, N.J.; Hemingway, D.M. Prognostic significance of elevated endothelin-1 levels in patients with colorectal cancer. Int. J. Biol. Markers 2004, 19, 32–37. [Google Scholar] [PubMed]
- Hohla, F.; Schally, A.V. Targeting gastrin releasing peptide receptors: New options for the therapy and diagnosis of cancer. Cell Cycle 2010, 9, 1738–1741. [Google Scholar] [CrossRef] [PubMed]
- Fosslien, E. Cardiovascular complications of non-steroidal anti-inflammatory drugs. Ann. Clin. Lab. Sci. 2005, 35, 347–385. [Google Scholar] [PubMed]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Faure, M.; Voyno-Yasenetskaya, T.A.; Bourne, H.R. CAMP and β γ subunits of heterotrimeric G proteins stimulate the mitogen-activated protein kinase pathway in COS-7 cells. J. Biol. Chem. 1994, 269, 7851–7854. [Google Scholar] [PubMed]
- Daub, H.; Wallasch, C.; Lankenau, A.; Herrlich, A.; Ullrich, A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997, 16, 7032–7044. [Google Scholar] [CrossRef] [PubMed]
- Zwick, E.; Wallasch, C.; Daub, H.; Ullrich, A. Distinct calcium-dependent pathways of epidermal growth factor receptor transactivation and PYK2 tyrosine phosphorylation in PC12 cells. J. Biol. Chem. 1999, 274, 20989–20996. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, S.; Iwasaki, H.; Ueno, H.; Frank, G.D.; Motley, E.D.; Eguchi, K.; Marumo, F.; Hirata, Y.; Inagami, T. Intracellular signaling of angiotensin II-induced p70 S6 kinase phosphorylation at Ser(411) in vascular smooth muscle cells. Possible requirement of epidermal growth factor receptor, Ras, extracellular signal-regulated kinase, and Akt. J. Biol. Chem. 1999, 274, 36843–36851. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Di Fiore, P.P.; Pierce, J.H.; Kraus, M.H.; Segatto, O.; King, C.R.; Aaronson, S.A. ErbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science 1987, 237, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Gerald, W.L.; Li, A.R.; Pan, Q.; Lal, P.; Ladanyi, M.; Chen, B. EGFR gene amplification in breast cancer: Correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod. Pathol. 2005, 18, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Scambia, G.; Panici, P.B.; Battaglia, F.; Ferrandina, G.; Baiocchi, G.; Greggi, S.; de Vincenzo, R.; Mancuso, S. Significance of epidermal growth factor receptor in advanced ovarian cancer. J. Clin. Oncol. 1992, 10, 529–535. [Google Scholar] [PubMed]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, S9–S15. [Google Scholar] [CrossRef]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar, H.; Basbaum, C. Platelet-activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat. Med. 2002, 8, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar, H.; Li, D.; Gallup, M.; Sidhu, S.; Drori, E.; Basbaum, C. Tobacco smoke-induced lung cell proliferation mediated by tumor necrosis factor α-converting enzyme and amphiregulin. J. Biol. Chem. 2003, 278, 26202–26207. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.C.; Liu, X.; Li, Y.; Covington, M.; Wynn, R.; Huber, R.; Hillman, M.; Yang, G.; Ellis, D.; Marando, C.; et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol. Ther. 2006, 5, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Ye, Z.Y.; Li, L.; Zhao, Z.S.; Shao, Q.S.; Tao, H.Q. ADAM 10 is associated with gastric cancer progression and prognosis of patients. J. Surg. Oncol. 2011, 103, 116–123. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.R.; Akl, P.; Samaratunga, H.; Herington, A.C.; Odorico, D.M. Expression of the disintegrin metalloprotease, ADAM-10, in prostate cancer and its regulation by dihydrotestosterone, insulin-like growth factor I, and epidermal growth factor in the prostate cancer cell model LNCaP. Clin. Cancer Res. 2004, 10, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Feldinger, K.; Generali, D.; Kramer-Marek, G.; Gijsen, M.; Ng, T.B.; Wong, J.H.; Strina, C.; Cappelletti, M.; Andreis, D.; Li, J.L.; et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget 2014, 5, 6633–6646. [Google Scholar] [CrossRef] [PubMed]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Ullrich, A. EGFR signal transactivation in cancer cells. Biochem. Soc. Trans. 2003, 31, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Okuma, Y.; Hosomi, Y.; Nagamata, M.; Yamada, Y.; Sekihara, K.; Kato, K.; Hishima, T.; Okamura, T. Clinical outcomes after first-line EGFR inhibitor treatment for patients with NSCLC, EGFR mutation, and poor performance status. Anticancer Res. 2013, 33, 5057–5064. [Google Scholar] [PubMed]
- Bhola, N.E.; Grandis, J.R. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front. Biosci. 2008, 13, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Krysan, K.; Reckamp, K.L.; Dalwadi, H.; Sharma, S.; Rozengurt, E.; Dohadwala, M.; Dubinett, S.M. Prostaglandin E2 activates mitogen-activated protein kinase/ERK pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 2005, 65, 6275–6281. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Bhola, N.E.; Zhang, Q.; Contrucci, S.C.; Wentzel, A.L.; Freilino, M.L.; Gooding, W.E.; Siegfried, J.M.; Chan, D.C.; Grandis, J.R. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006, 66, 11831–11839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bhola, N.E.; Lui, V.W.Y.; Siwak, D.R.; Thomas, S.M.; Gubish, C.T.; Siegfried, J.M.; Mills, G.B.; Shin, D.; Grandis, J.R. Antitumor mechanisms of combined gastrin-releasing peptide receptor and epidermal growth factor receptor targeting in head and neck cancer. Mol. Cancer Ther. 2007, 6, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Corps, A.N.; Rees, L.H.; Brown, K.D. A peptide that inhibits the mitogenic stimulation of Swiss 3T3 cells by bombesin or vasopressin. Biochem. J. 1985, 231, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T.; Jones, S.W.; Folkers, K.; Gardner, J.D. A Synthetic peptide that is a bombesin receptor antagonist. Nature 1984, 309, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.H.; Casley, D.J.; Nayler, W.G. The inhibitory effect of D-Arg1, D-Phe, D-Try7, 9, Leu11 substance P on endothelin-1 binding sites in rat cardiac membranes. Biochem. Biophys. Res. Commun. 1991, 179, 130–133. [Google Scholar] [CrossRef]
- Wang, J.; He, L.; Combs, C.A.; Roderiquez, G.; Norcross, M.A. Dimerization of CXCR4 in living malignant cells: Control of cell migration by a synthetic peptide that reduces homologous CXCR4 interactions. Mol. Cancer Ther. 2006, 5, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Covic, L.; Gresser, A.L.; Talavera, J.; Swift, S.; Kuliopulos, A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc. Natl. Acad. Sci. USA 2002, 99, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Covic, L.; Misra, M.; Badar, J.; Singh, C.; Kuliopulos, A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat. Med. 2002, 8, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Rajagopal, K.; Whalen, E.J. New roles for β-arrestins in cell signaling: Not just seven-transmembrane receptors. Mol. Cell 2006, 24, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by β-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPCR | Role | Stem Cell Type | Reference |
|---|---|---|---|
| Gαs | Promotes proliferation and pluripotency | Mouse ESCs | [11] |
| Gαi | Regulates morphology and cellular organization | Human iPSCs | [10] |
| Gαi/o | Regulates Wnt signaling activation | Human ESCs | [42,44] |
| Fzd7 | Inhibition of Fzd7 induces significant morphological alterations with loss of pluripotency gene Oct4 | Human ECSs | [57] |
| GPR84 | Promotes β-catenin signaling and LSC maintenance | AML LSCs | [70] |
| Gαq | Enhances β-catenin signaling contributing to maintenance of fully-developed AML | AML LSCs | [71] |
| Lgr5 | Potentiates Wnt signaling, drives migration and metastasis | Colon CSCs | [81,86,88] |
| Lgr6 | Drives stem cell self-renewal | Hair follicle epidermal stem cells | [66] |
| Lgr4 | Enhances Wnt signaling, promotes CSC self-renewal and maintenance | Prostate CSCs, mammary CSCs and AML LSCs | [66,92,98,100] |
| EP1 | Regulates β-catenin driven self-renewal and stem cell frequency | AML LSCs | [65,101] |
| EP4 | Enhances metastasis and tumorigenicity | Breast CSCs | [103] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lynch, J.R.; Wang, J.Y. G Protein-Coupled Receptor Signaling in Stem Cells and Cancer. Int. J. Mol. Sci. 2016, 17, 707. https://doi.org/10.3390/ijms17050707
Lynch JR, Wang JY. G Protein-Coupled Receptor Signaling in Stem Cells and Cancer. International Journal of Molecular Sciences. 2016; 17(5):707. https://doi.org/10.3390/ijms17050707
Chicago/Turabian StyleLynch, Jennifer R., and Jenny Yingzi Wang. 2016. "G Protein-Coupled Receptor Signaling in Stem Cells and Cancer" International Journal of Molecular Sciences 17, no. 5: 707. https://doi.org/10.3390/ijms17050707
APA StyleLynch, J. R., & Wang, J. Y. (2016). G Protein-Coupled Receptor Signaling in Stem Cells and Cancer. International Journal of Molecular Sciences, 17(5), 707. https://doi.org/10.3390/ijms17050707