
A Comparative Analysis of Genetic and Epigenetic Events of Breast and Ovarian Cancer Related to Tumorigenesis



Abstract
:
1. Classification of Breast and Ovarian Cancer
2. Comparison of the Genetic and microRNA Environments of Breast and Ovarian Cancer
3. Comparison of the Respective MicroRNA Environments
4. Epigenetic Comparison of Breast and Ovarian Cancer
5. Role of Stroma in Breast and Ovarian Cancer
6. Current and Future Treatment of Breast and Ovarian Cancer
7. Drug Resistance
8. Inferences from a Comparison of Breast and Ovarian Cancer: An Epigenetic Link
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AKR1B1 | Aldo-keto reductase family 1, member B1 |
| APC | Adenomatous polyposis coli |
| ARHI | Aplysia ras homology member I (GTP binding protein Di-RAS3) |
| ARID3B | AT-rich interactive domain-containing protein 3B |
| CBFB | Core-binding factor subunit beta |
| CDH1 | Cadherin-1 |
| CDK | Cyclin kependent kinase |
| CDKL2 | Cyclin-dependent kinase-like 2 |
| CREBBP | CREB-binding protein |
| DBX1 | developing brain homeobox protein 1 |
| DNMT | DNA methyltransferase |
| DNMTi | DNA methyltransferase inhibitor |
| EGF | Epidermal growth factor |
| EMT | Epithelial mesenchymal transition |
| ER | Estrogen receptor |
| ER- | Estrogen receptor negative |
| EZH2 | Enhancer of zeste homolog 2 |
| FANCF | Fanconi anemia complementation group F |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FRAP1 | FK506-binding protein 12-rapamycin-associated protein 1 (Mammalian target of rapamycin, MTOR) |
| GABRA6 | Gamma-aminobutyric acid receptor subunit alpha-6 |
| GSTP1 | Glutathione S-transferase P |
| HDACi | Histone deacetylase inhibitor |
| HGS-OvCa | high-grade serous ovarian cancer |
| HIF | Hypoxia induced factor |
| HK1 | Hexokinase-1 |
| IRF2BP2 | Interferon regulatory factor 2-binding protein 2 |
| IRF4 | interferon regulatory factor 4 |
| KMT2C | Lysine N-methyltransferase 2C (Mixed-lineage leukemia protein 3, MLL3) |
| MAP3K1 | (Mitogen-Activated Protein Kinase Kinase Kinase 1 |
| MDR | Multidrug resistant |
| MECOM | MDS1 and EVI1 complex locus protein EVI1 |
| MGMT | O-6-Methylguanine-DNA methyltransferase |
| miR | microRNA |
| miRNA | microRNA |
| MLH1 | MutL homolog 1 |
| MMP9 | Matrix metallopeptidase 9 |
| NEUROD1 | Neuronal differentiation 1 |
| NF1 | Neurofibromin 1 |
| PAX8 | Paired box gene 8 |
| PFKP | Phosphofructokinase, platelet |
| PIK3 | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| PTEN | Phosphatase and tensin homolog |
| PTPRD | Protein tyrosine phosphatase, receptor type D |
| PVRL2 | Poliovirus receptor-related 2 |
| RASSF1A | Ras association domain-containing protein 1 |
| RB1 | Retinoblastoma 1 |
| RUNX1 | Runt-related transcription factor 1 |
| SF3B1 | Splicing factor 3B subunit 1 |
| TERT | Telomerase reverse transcriptase |
| TGF | Transforming growth factor |
| TNBC | triple negative basal cell breast cancer |
| TxF | Transcription factor |
| USF2 | Upstream stimulatory factor 2 |
| ZEB | Zinc finger E-box-binding homeobox 1 |
| ZMYND8 | Protein kinase C-binding protein 1 |
References
- Langerød, A.; Zhao, H.; Borgan, Ø.; Nesland, J.M.; Bukholm, I.R.; Ikdahl, T.; Kåresen, R.; Børresen-Dale, A.-L.; Jeffrey, S.S. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Burgess, M.; Puhalla, S. BRCA 1/2-Mutation related and sporadic breast and ovarian cancers: More alike than different. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [Green Version]
- Sadeqzadeh, E.; de Bock, D.E.; Thorne, R.F. Sleeping giants: Emerging roles for cadherins in health and disease. Med. Res. Rev. 2014, 34, 190–221. [Google Scholar] [CrossRef] [PubMed]
- Oeschger, F.M.; Wang, W.; Lee, S.; Garcia-Moreno, F.; Goffinet, A.M.; Arbones, M.I.; Rakic, S.; Molnar, Z. Gene expression analysis of the embryonic subplate. Cereb. Cortex 2012, 6, 1343–1359. [Google Scholar] [CrossRef] [PubMed]
- Ekumi, K.M.; Paculova, H.; Lenasi, T.; Popichalova, V.; Bosken, C.A.; Rybarikova, J.; Bryja, V.; Geyer, M.; Blazek, D.; Barboric, M. Ovarian carcinoma CKD12 mutations misregulate expression of DNA repair genes via Cdk12/CycK complex. Nucleic Acids Res. 2015, 5, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Koff, A.; Giordano, A.; Desai, D.; Yamashita, K.; Harper, J.W.; Elledge, S.; Nishimoto, T.; Morgan, D.O.; Franza, B.R.; Roberts, J.M. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science 1992, 257, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.; Wilson, M.; Gonzalez, R.; Zhang, Y.; Perkins, A.S. The role of EVI1 in myeloid malignancies. Blood Cells Mol. Dis. 2014, 53, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Chiu, L.-Y.; Cox, B.; Aymard, F.; Clouaire, T.; Leung, J.W.; Cammarata, M.; Perez, M.; Agarwal, P.; Brodbelt, J.S.; et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 2015, 29, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Ling, F.; Kang, B.; Sun, X. Id proteins: Small molecules, mighty regulators. Curr. Top. Dev. Biol. 2014, 110, 189–216. [Google Scholar] [PubMed]
- Mittag, J.; Winterhager, E.; Bauer, K.; Grummer, R. Congenital hypothyroid femail pax8-deficient mice are infertile despite thyroid hormone replacement therapy. Endocrinology 2007, 148, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Dydensborg, A.B.; Rose, A.A.N.; Wilson, B.J.; Grote, D.; Paquet, M.; Giguère, V.; Siegel, P.M.; Bouchard, M. GATA3 inhibits breast cancer growth and pulmonary breast cancer metastasis. Oncogene 2009, 28, 2634–2642. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.J.; Dingwall, A.K. The cancer COMPASS: Navigating the functions of MLL complexes in cancer. Cancer Genet. 2015, 208, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.; Grothey, E.; Couch, F.J. The role of Tbx2 and Tbx3 in mammary development and tumorigenesis. J. Mammary Gland Biol. Neoplasia 2004, 2, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Melko, M.; Nguyen, L.S.; Shaw, M.; Jolly, L.; Bardoni, B.; Gecz, J. Loss of FMR2 further emphasizes the link between deregulation of immediate early response genes FOS and JUN and intellectual disability. Hum. Mol. Genet. 2013, 22, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Prat, J. New insights into ovarian cancer pathology. Ann. Oncol. 2012, 23, x111–x117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Helland, A.; Holm, R.; Skomedal, H.; Abeler, V.M.; Danielsen, H.E.; Tropé, C.G.; Børresen-Dale, A.-L.; Kristensen, G.B. TP53 mutations in early-stage ovarian carcinoma, relation to long-term survival. Br. J. Cancer 2004, 90, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kringen, P.; Kristensen, G.B.; Holm, R.; Baekelandt, M.M.O.; Olivier, M.; Skomedal, H.; Hainaut, P.; Tropé, C.G.; Abeler, V.M.; et al. Effect of the codon 72 polymorphism (c.215G>C, p.Arg72Pro) in combination with somatic sequence variants in the TP53 gene on survival in patients with advanced ovarian carcinoma. Hum. Mutat. 2004, 24, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.; Beiner, M.; Lubinski, J.; Lynch, H.T.; Moller, P.; Rosen, B.; Murphy, J.; Ghadirian, P.; Friedman, E.; Foulkes, W.D.; et al. Hereditary Ovarian Cancer Clinical Study Group. Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 mutation. JAMA 2006, 296, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, C.R.; Levine, D.A.; Tang, L.H.; Allen, P.J.; Jarnagin, W.; Brennan, M.F.; Offit, K.; Robson, M.E. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J. Clin. Oncol. 2009, 27, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Tian, L.; Kähkönen, M.; Schwartzentruber, J.; Kircher, M.; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium; Majewski, J.; Dyment, D.A.; Innes, A.M.; et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Severson, T.M.; Peeters, J.; Majewski, I.; Michaut, M.; Bosma, A.; Schouten, P.C.; Chin, S.F.; Pereira, B.; Goldgraben, M.A.; Bismeijer, T.; et al. BRCA1-line signature in triple negative breast cancer: Molecular and clinical characterization reveals subgroups with therapeutic potential. Mol. Oncol. 2015, 9, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Lips, E.H.; Laddach, N.; Savola, S.P.; Vollebergh, M.A.; Oonk, A.M.; Imholz, A.L.; Wessels, L.F.; Wesseling, J.; Nederlof, P.M.; Rodenhuis, S. Quantitative copy number analysis by Multiplex Ligation-dependent Probe Amplification (MLPA) of BRCA1-associated breast cancer regions identifies BRCAness. Breast Cancer Res. 2011, 13, R107. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, A.K.; Creighton, C.J.; Yu, Z.; Zhu, H.; Gunaratne, P.H.; Reid, J.G.; Olokpa, E.; Itamochi, H.; Ueno, N.T.; Hawkins, S.M.; et al. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol. Endocrinol. 2010, 24, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.-G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Derfoul, A.; Juan, A.H.; Difilippantonio, M.J.; Palanisamy, N.; Ried, T.; Sartorelli, V. Decreased microRNA-214 levels in breast cancer cells coincides with increased cell proliferation, invasion and accumulation of the Polycomb Ezh2 methyltransferase. Carcinogenesis 2011, 32, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- O’Day, E.; Lal, A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010, 12, 201. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. microRNA-antagonism regulates breast cancer stemness and metastasis via TET family dependent chromatin remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Jiao, Y.; Zhu, Y.; Wang, Y.; Zhu, J.; Cui, X.; Liu, Y.; He, Y.; Park, E.-Y.; Zhang, H.; et al. MicroRNA 34c gene down-regulation via DNA methylation promotes self-renewal and epithelial-mesenchymal transition in breast tumor-initiating cells. J. Biol. Chem. 2012, 287, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kong, W.; He, L.; Zhao, J.-J.; O’Donnell, J.D.; Wang, J.; Wenham, R.M.; Coppola, D.; Kruk, P.A.; Nicosia, S.V.; et al. MicroRNA expression profiling in human ovarian cancer: MiR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008, 68, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Giannakakis, A.; Sandaltzopoulos, R.; Greshock, J.; Liang, S.; Huang, J.; Hasegawa, K.; Li, C.; O’Brien-Jenkins, A.; Katsaros, D.; Weber, B.L.; et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol. Ther. 2008, 7, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Laios, A.; O’Toole, S.; Flavin, R.; Martin, C.; Kelly, L.; Ring, M.; Finn, S.P.; Barrett, C.; Loda, M.; Gleeson, N.; et al. Potential role of miR-9 and miR-223 in recurrent ovarian cancer. Mol. Cancer 2008, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Furneaux, H.; White, B.A. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol. Endocrinol. 2007, 21, 1132–1147. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Huang, R.; Wang, Z. Potential role of miR-100 in cancer diagnosis, prognosis, and therapy. Tumor Biol. 2015, 36, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wei, W.; Sun, Y.; Gao, J.; Wang, Q.; Zheng, J. MicroRNA-9 promotes tumorigenesis and mediates sensitivity to cisplatin in primary epithelial ovarian cancer. Tumor Biol. 2015, 36, 6867–6873. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Gao, Y.; Zhou, J.; Wang, J.; Zheng, F.; Guo, F.; Chang, A.; Li, X.; Wang, B. miR-233 regulates adipogenic and osteogenic differentiation of mesenchymal stem cells through a C/EBPs/miR-233/FGFR2 regualtory feedback loop. Stem Cells 2015, 33, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, M.; Mir, R.; Das, J.; Ahmad, I.; Javid, J.; Yadav, P.; Masroor, M.; Ahmad, S.; Ray, P.C.; Saxena, A. Expression of serum miR-200a, miR-200b, and miR-200c as candidate biomarkers in epithelial ovarian cancer and their association with clinicopathological features. Clin. Transl. Oncol. 2015, 17, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Mukohyama, J.; Nakamura, S.; Minami, H. MicroRNA regulation of human breast cancer stem cells. J. Clin. Med. 2016, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Guo, Y.; Chen, Z.; Wang, Y.; Yang, C.; Dudas, A.; Du, Z.; Liu, W.; Zou, Y.; Szabo, E.; et al. miR-203 functions as a tumor suppressor by inhibiting epithelial to mesenchymal transition in ovarian cancer. J. Cancer Sci. Ther. 2015, 7, 34–43. [Google Scholar] [PubMed]
- Zhang, S.; Lu, Z.; Unruh, A.K.; Ivan, C.; Baggerly, K.A.; Calin, G.A.; Li, Z.; Bast, R.C.; Le, X. Clinically relevant microRNAs in ovarian cancer. Mol. Cancer Res. 2015, 13, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.D.C.; Dahl, R.; Kruichak, J.N.; Hudson, L.G. The epidermal growth factor receptor responsive miR-125a represses mesenchymal morphology in ovarian cancer cells. Neoplasia 2009, 11, 1208–1215. [Google Scholar] [CrossRef]
- Byler, S.; Sarkar, S. Do epigenetic drug treatments hold the key to killing cancer progenitor cells? Epigenomics 2014, 6, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kimball, S.; Liu, H.; Holowatyj, A.; Yang, Z.Q. Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget 2014, 6, 2466–2482. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.Y.; Lei, P.J.; Zhang, X.; Zheng, J.Y.; Wang, H.Y.; Zhao, J.; Li, Y.M.; Ye, M.; Li, L.; Wei, G.; et al. Global histone modification profiling reveals the epigenomic dynamics during malignant transformation in a four-stage breast cancer model. Clin. Epigenet. 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D.J.; Shah, J.S.; Cole, A.J. Histones and Their Modifications in Ovarian Cancer—Drivers of Disease and Therapeutic Targets. Front. Oncol. 2014, 4, 144. [Google Scholar] [PubMed]
- Rhie, S.K.; Hazelett, D.J.; Coetzee, S.G.; Yan, C.; Noushmehr, H.; Coetzee, G.A. Nucleosome positioning and histone modifications define relationships between regulatory elements and nearby gene expression in breast epithelial cells. BMC Genom. 2014, 15, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, A.; Cross, N.C.P. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Kron, K.J.; Bailey, S.D.; Lupien, M. Enhancer altertions in cancer: A source for a cell identity crisis. Genome Med. 2014, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Niederriter, A.R.; Varshney, A.; Parker, S.C.; Martin, D.M. Super enhancers in cancers, complex diseases, and developmental disorders. Genes (Basel) 2015, 4, 1183–1200. [Google Scholar] [CrossRef] [PubMed]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, J.I.; Zhao, K.; et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maurano, M.T.; Qu, H.; Varley, K.E.; Gertz, J.; Pauli, F.; Lee, K.; Canfield, T.; Weaver, M.; Sandstrom, R.; et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012, 22, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Goldgar, S.; Byler, S.; Rosenthal, S.; Heerboth, S. Demethylation and re-expression of epigenetically silenced tumor suppressor genes: Sensitization of cancer cells by combination therapy. Epigenomics 2013, 5, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef] [PubMed]
- Koukoura, O.; Spandidos, D.A.; Daponte, A.; Sifakis, S. DNA methylation profiles in ovarian cancer: Implication in diagnosis and therapy (Review). Mol. Med. Rep. 2014, 10, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lavaud, P.; Andre, F. Strategies to overcome trastuzumab resistance in HER2-overexpressing breast cancers: Focus on new data from clinical trials. BMC Med. 2014, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Deftereos, G. DNA hypermethylation, Her-2/neu overexpression and p53 mutations in ovarian carcinoma. Gynecol. Oncol. 2008, 111, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, H.; Millinger, S.; Goebel, G.; Müller-Holzner, E.; Marth, C.; Laird, P.W.; Widschwendter, M. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: Association with HER-2/neu status in primary breast cancer. Cancer Res. 2006, 66, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, B.M.; Wingren, S.; Motlagh, P.B.; Nilsson, T.K. Whole genome DNA methylation signature of HER2-positive breast cancer. Epigenetics 2014, 9, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Rodenhiser, D.I.; Andrews, J.; Kennette, W.; Sadikovic, B.; Mendlowitz, A.; Tuck, A.B.; Chambers, A.F. Epigenetic mapping and functional analysis in a breast cancer metastasis model using whole-genome promoter tiling microarrays. Breast Cancer Res. 2008, 10. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Herman, J.G.; Lapidus, R.G.; Chopra, H.; Xu, R.; Jarrard, D.F.; Isaacs, W.B.; Pitha, P.M.; Davidson, N.E.; Baylin, S.B. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995, 55, 5195–5199. [Google Scholar] [PubMed]
- Do, S.-I.; Ko, E.; Kang, S.Y.; Lee, J.E.; Nam, S.J.; Cho, E.Y.; Kim, D.-H. Aberrant DNA methylation of integrin α4 in human breast cancer. Tumour Biol. 2014, 35, 7079–7084. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Kersch, C.; Beeson, K.A.; Wu, Y.J.; Muldoon, L.L.; Neuwelt, E.A. Interactions between αv-integrin and HER2 and their role in the invasive phenotype of breast cancer cells in vitro and in rat brain. PLoS ONE 2015, 10, e0131842. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Abujamra, A.L.; Loew, J.E.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011, 31, 2723–2732. [Google Scholar] [PubMed]
- Zuo, T.; Liu, T.-M.; Lan, X.; Weng, Y.-I.; Shen, R.; Gu, F.; Huang, Y.-W.; Liyanarachchi, S.; Deatherage, D.E.; Hsu, P.-Y.; et al. Epigenetic silencing mediated through activated PI3K/AKT signaling in breast cancer. Cancer Res. 2011, 71, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Keyomarsi, K.; Gonzales, F.A.; Velicescu, M.; Jones, P.A. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res. 2000, 28, 2108–2113. [Google Scholar] [CrossRef] [PubMed]
- Samorodnitsky, E.; Ghosh, E.; Mazumder, S.; Sarkar, S. Methylation by DNMT1 is more Efficient in Chronic Lymphocytic Lymphoma Cells than in Normal Cells. J. Proteom. Bioinform. 2014, 1. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, S.; Song, E.; Liu, S. The roles of ncRNAs and histone-modifiers in regulating breast cancer stem cells. Protein Cell 2016, 7, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Rosen, D.G.; Zhang, Z.; Bast, R.C.; Mills, G.B.; Colacino, J.A.; Mercado-Uribe, I.; Liu, J. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 16472–16477. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gauthier, A.; Postma, M.J.; Ribassin-Majed, L.; Largeron, N.; Bresse, X. A critical review of cost-effectiveness analyses of vaccinating males against human papillomavirus. Hum. Vaccines Immunother. 2013, 9, 2285–2295. [Google Scholar] [CrossRef]
- Matei, D.; Sill, M.W.; Lankes, H.A.; DeGeest, K.; Bristow, R.E.; Mutch, D.; Yamada, S.D.; Cohn, D.; Calvert, V.; Farley, J.; et al. Activity of sorafenib in recurrent ovarian cancer and primary peritoneal carcinomatosis: A gynecologic oncology group trial. J. Clin. Oncol. 2011, 29, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Longacre, M.; Tatur, N.; Heerboth, S.; Lapinska, K. Encyclopedia of Analytical Chemistry; Histone Decatylases (HDACs): Function, Mechanism & Inhibition; Wiley Online Library: London, UK, 2014; pp. 1–9. [Google Scholar]
- Chang, H.G.; Kim, S.J.; Chung, K.-W.; Noh, D.-Y.; Kwon, Y.; Lee, E.S.; Kang, H.-S. Tamoxifen-resistant breast cancers show less frequent methylation of the estrogen receptor beta but not the estrogen receptor alpha gene. J. Mol. Med. 2005, 83, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Glasspool, R.M.; Teodoridis, J.M.; Brown, R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br. J. Cancer 2006, 94, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Strathdee, G.; Vass, J.K.; Oien, K.A.; Siddiqui, N.; Curto-Garcia, J.; Brown, R. Demethylation of the MCJ gene in stage III/IV epithelial ovarian cancer and response to chemotherapy. Gynecol. Oncol. 2005, 97, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Gifford, G.; Paul, J.; Vasey, P.A.; Kaye, S.B.; Brown, R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin. Cancer Res. 2004, 10, 4420–4426. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.E.; Nephew, K.P. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS ONE 2014, 9, e87455. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Unterberg, M.; Koschmieder, S.; zur Stadt, U.; Brunnberg, U.; Verbeek, W.; Büchner, T.; Berdel, W.E.; Serve, H.; Müller-Tidow, C. DNA methylation of tumor suppressor genes in clinical remission predicts the relapse risk in acute myeloid leukemia. Cancer Res. 2007, 67, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.; Huang, T.H.-M.; Brown, R.; Nephew, K.P. The epigenetics of ovarian cancer drug resistance and resensitization. Am. J. Obstet. Gynecol. 2004, 191, 1552–1572. [Google Scholar] [CrossRef] [PubMed]
- Mataga, M.A.; Rosenthal, S.; Heerboth, S.; Devalapalli, A.; Kokolus, S.; Evans, L.R.; Longacre, M.; Housman, G.; Sarkar, S. Anti-breast cancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Res. 2012, 32, 2523–2529. [Google Scholar] [PubMed]
- Lapinska, K.; Housman, G.; Heerboth, S.; Longacre, M.; Sarkar, S. Anticancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Res. 2012, 32, 2523–2529. [Google Scholar]
- Fujji, S.; Luo, R.Z.; Yuan, J.; Kadota, M.; Oshimura, M.; Dent, S.R.; Kondo, Y.; Issa, J.J.; Bast, R.C.; Yu, Y. Reactivation of the silenced and imprinted alleles of ARHI is associated with increased histone H3 acetylation and decreased histone H3 lysine 9 methylation. Hum. Mol. Genet. 2003, 12, 1791–1800. [Google Scholar] [CrossRef]
- Frew, A.J.; Lindemann, R.K.; Martin, B.P.; Clarke, C.J.P.; Sharkey, J.; Anthony, D.A.; Banks, K.-M.; Haynes, N.M.; Gangatirkar, P.; Stanley, K.; et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc. Natl. Acad. Sci. USA 2008, 105, 11317–11322. [Google Scholar] [CrossRef] [PubMed]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non–small cell lung cancer. Cancer Discov. 2011. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Faller, D.T. T-oligos inhibit growth and induce apoptosis in human ovarian cancer cells. Oligonucleotides 2011, 21, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Alfraidi, A.; Sacco, K.; Alshaker, H.; Muhammad, A.; Monzon, L. Histone deacetylases as new therapy targets for platinum-resistant epithelial ovarian cancer. J. Cancer Res. Clin. Oncol. 2015, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyparaki, M.T.; Papavassiliou, A.G. Epigenetic pathways offer targets for ovarian cancer treatment. Clin. Ovarian Other Gynecol. Cancer 2014, 4, 71. [Google Scholar] [CrossRef]
- Dong, Y.; Batra, J.; Anand, K.S.; Bapat, S.; Clements, A.J. Transforming the future of treatment for ovarian cancer. Clin. Exp. Pharmacol. 2014, 4, 3. [Google Scholar]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Byler, S.; Goldgar, S.; Heerboth, S.; Leary, M.; Housman, G.; Moulton, K.; Sarkar, S. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014, 34, 1071–1077. [Google Scholar] [PubMed]




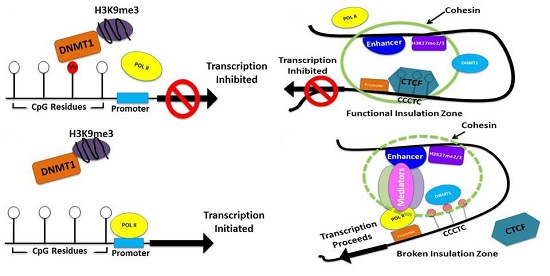{kind=link}
{kind=link}
{kind=link}
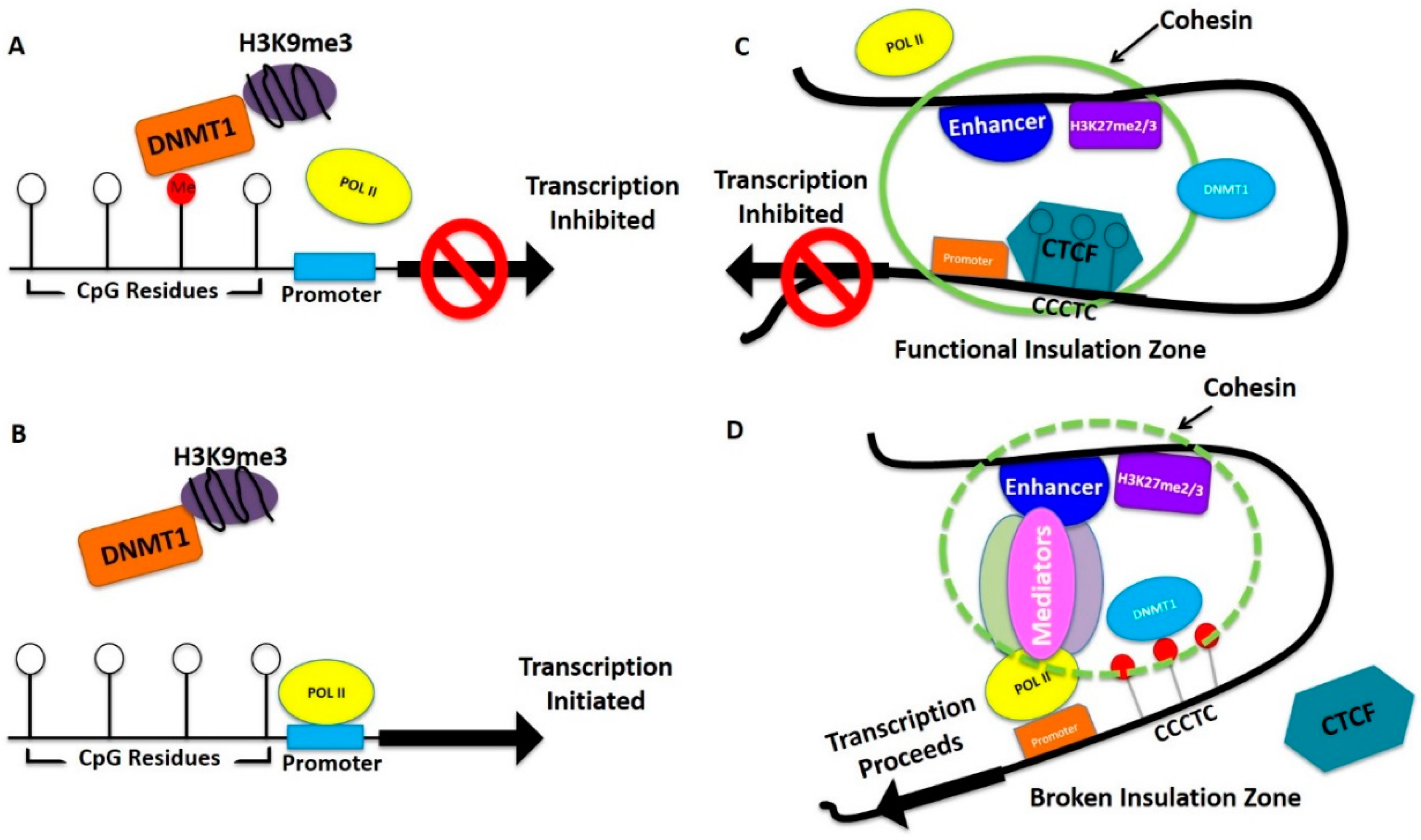{kind=link}
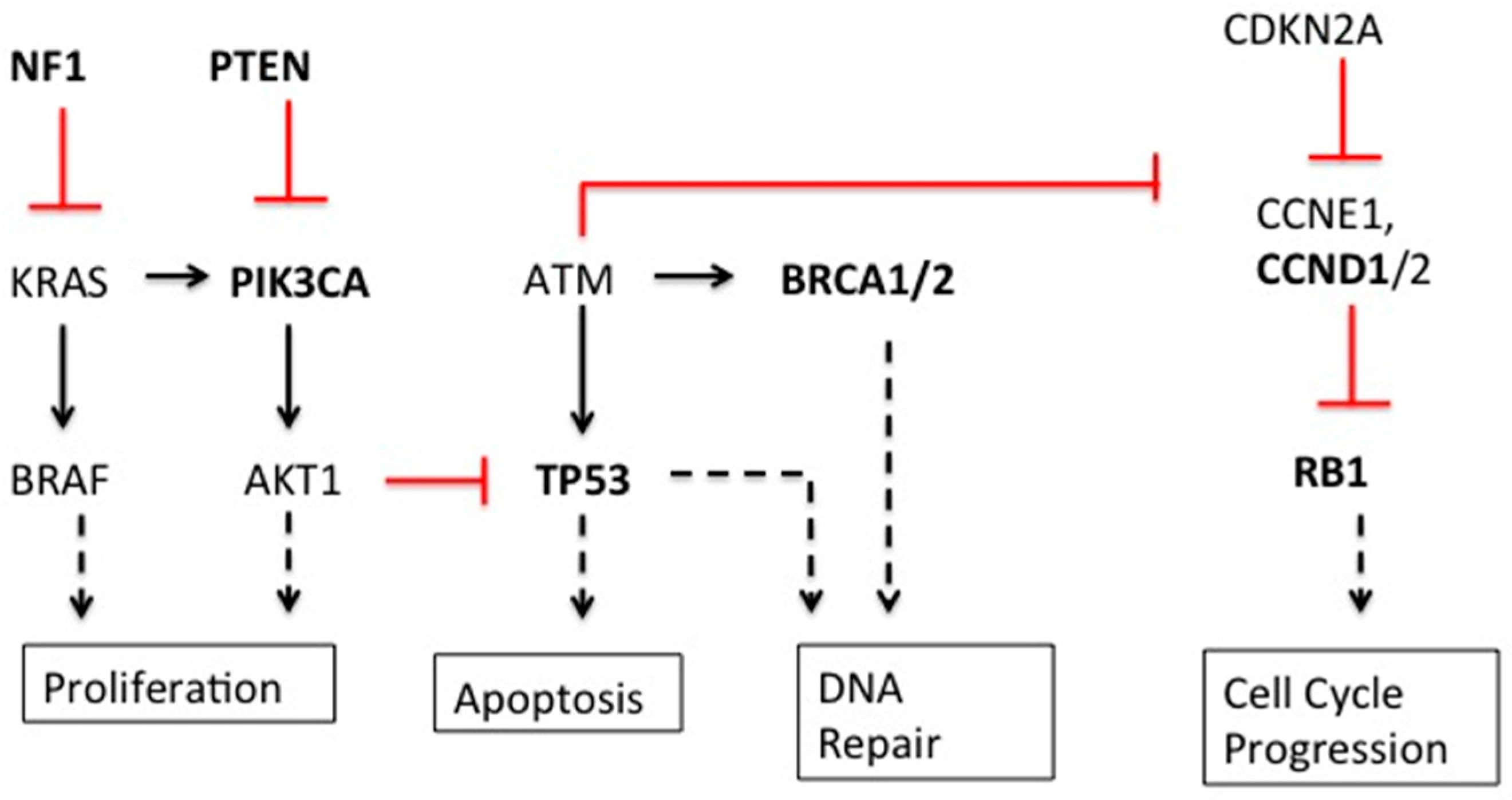
| Gene | Type | Gene Function | Breast | Ovarian |
|---|---|---|---|---|
| BRCA1/2 [3,4,5] | Mutation | DNA homologous recombination repair | Yes | Yes |
| TP53 [1,3,5] | Mutation | Cell cycle checkpoint | Yes | Yes |
| RB1 [3,5] | Mutation/Deletion | Cell cycle regulator | Yes | Yes |
| NF1 [3,5] | Mutation/Deletion | Negative regulator cell division via Ras inhibition | Yes | Yes |
| FAT3 [3,5,6] | Mutation | Central nervous system development | Yes | Yes |
| CSMD3 [3,5,7] | Mutation, Copy Number | Development | - | Yes |
| GABRA6 [5] | Mutation | GABA receptor, neurons | - | Yes |
| CDK12 [5,8] | Mutation | RNA splicing regulation | - | Yes |
| BRAF [5] | Mutation | Proto-oncogene, cell growth signals | - | Yes |
| PIK3CA [3,5] | Mutation | Cell growth, Catalytic subunit of PI3k, signaling cascades including activation of Akt | Yes | Yes |
| KRAS [5] | Mutation | Cell growth, Signal propagation including growth factor and PI3k signals | - | Yes |
| NRAS [5] | Mutation | Cell growth, Signal propagation including growth factor and PI3k signals | - | Yes |
| CCNE1 [5,9] | Copy Number Amplification | Cycle E1—cell cycle regulation | - | Yes |
| MYC [3,5] | Copy Number Amplification | Txn factor, involved in cell cycle progression and apoptosis | Yes | Yes |
| MECOM [5,10] | Copy Number Amp | Differentiation, apoptosis, stem cell quiescence [6] | - | Yes |
| ZMYND8 [5,11] | Copy Number Amp | C-kinase receptor, possibly involved in DNA damage recognition [7] | - | Yes |
| IRF2BP2 [5] | Copy Number Amp | P53 target | - | Yes |
| ID4 [5,12] | Copy Number Amp | Transcription inhibition development, growth differentiation, senescence, apoptosis, angiogenesis | - | Yes |
| PAX8 [5,13] | Copy Number Amp | Development | - | Yes |
| TERT [5] | Copy Number Amp | Telomerase, genome stability | - | Yes |
| PTEN [3,5] | Deletion | cell cycle and apoptosis, possibly migration, adhesion, and angiogenesis | Yes | Yes |
| CREBBP [5] | Deletion | Cell cycle control | Yes | |
| AKT1 [3] | Apoptosis | Yes | - | |
| GATA3 [3,14] | Mutation | Differentiation of luminal cells, Estrogen Receptor pathway | Yes | - |
| CDH1 [3] | - | Cell adhesion, cell cycle regulation | Yes | - |
| MLL3/KMT2C [3,15] | - | Demethylation of H3K27, differentiation | Yes | - |
| MAP3K1 [3] | - | MAPK/ERK pathway-cell cycle | Yes | - |
| CDK1B [3] | - | Cell cycle progression | Yes | - |
| TBX3 [3,16] | Mutation | Mammary gland development | Yes | - |
| RUNX1 [3] | - | Development and differentiation, hematopoiesis | Yes | - |
| CBFB [3] | - | Development, stem-cell homeostasis | Yes | - |
| AFF2 [3,5,17] | - | Cell proliferation | Yes | Yes |
| PTPN22 [3] | - | Immune signaling, responsiveness of T and B cells | Yes | - |
| PTPRD [3] | - | Cell cycle, growth, differentiation | Yes | - |
| SF3B1 [3] | - | Splicing | Yes | - |
| CCND3 [3] | - | Cell cycle | Yes | - |
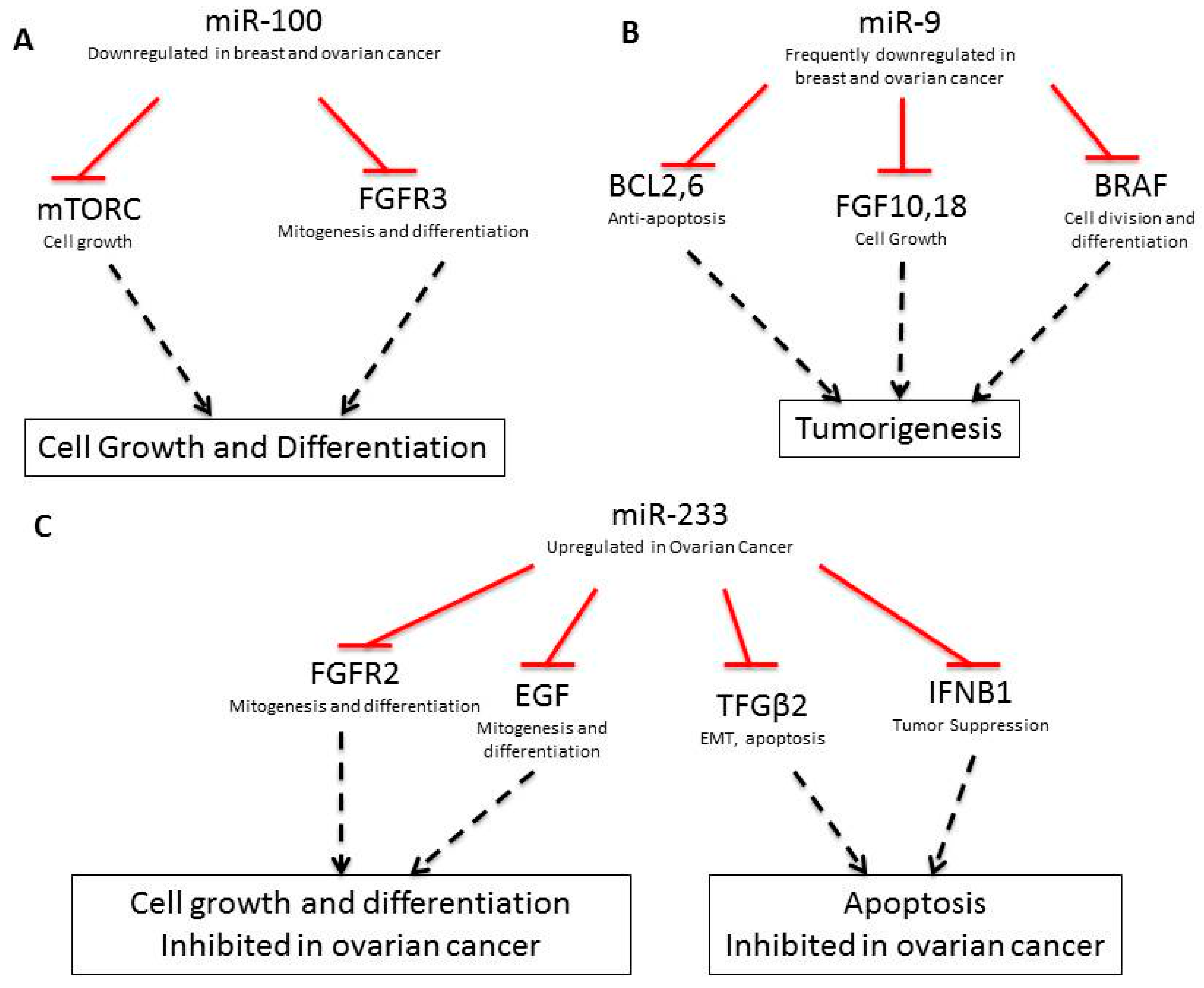
| miRNA | Up-/Downregulated | Gene Target | Gene Activity | Breast Cancer? | Ovarian Cancer? | Function |
|---|---|---|---|---|---|---|
| miR-100 [29] | Down | FRAP1/mTOR, FGFR3 [30] | - | Yes | Yes | Cell growth and survival |
| miR-9 [31] | Down | FGF18, FGF10, BCL2, BCL6, BRAF, CLDN14, CLDN6, SEPTIN10, ZNF, PVRL2, LASS4, BCL2, CLDN, FGF | - | Yes (miR-9-3) | Yes | Drug resistance |
| miR-214 [29,32] | Down | PTEN, EZH2 | Suppression | Yes | Yes | Cell survival, cisplatin resistance, Akt |
| miR-125a [33] | Down | HER2, ARI3B | Suppression | Yes | Yes | EMT |
| miR-125b [33] | Down | HER2 | Suppression | Yes | Yes | EMT |
| miR-22 [34,35] | - | miR-20 | - | Yes | - | Metastasis |
| miR-34c [36] | Down | - | - | - | - | EMT |
| miR-199a [37] | Down | - | - | - | Yes | |
| miR-200a [38] | Down(EMT)/Up(MET) | ZEB2 | Suppression | - | Yes | EMT |
| miR-200c [38] | Down(EMT)/Up(MET) | ZEB1/2 | Suppression | - | Yes | EMT |
| miR-146a | Up (variant allele) | BRCA1/2 | - | - | Yes | - |
| miR-210 [39] | Down (CNA) | E2F3 (TxF) | - | - | Yes | HIF |
| miR-233 [40] | Up | FGFR2, EGF, S100A3, KRAS, TGFΒ2, IFNBI, SPINKS, E2F1, SEPTIN6, MMP9, USF2 | - | - | - | Ras, integrin signal |
| miR-206 [33,41] | Up | ERα | Suppression | Yes | - | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longacre, M.; Snyder, N.A.; Housman, G.; Leary, M.; Lapinska, K.; Heerboth, S.; Willbanks, A.; Sarkar, S. A Comparative Analysis of Genetic and Epigenetic Events of Breast and Ovarian Cancer Related to Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 759. https://doi.org/10.3390/ijms17050759
Longacre M, Snyder NA, Housman G, Leary M, Lapinska K, Heerboth S, Willbanks A, Sarkar S. A Comparative Analysis of Genetic and Epigenetic Events of Breast and Ovarian Cancer Related to Tumorigenesis. International Journal of Molecular Sciences. 2016; 17(5):759. https://doi.org/10.3390/ijms17050759
Chicago/Turabian StyleLongacre, Mckenna, Nicole A. Snyder, Genevieve Housman, Meghan Leary, Karolina Lapinska, Sarah Heerboth, Amber Willbanks, and Sibaji Sarkar. 2016. "A Comparative Analysis of Genetic and Epigenetic Events of Breast and Ovarian Cancer Related to Tumorigenesis" International Journal of Molecular Sciences 17, no. 5: 759. https://doi.org/10.3390/ijms17050759
APA StyleLongacre, M., Snyder, N. A., Housman, G., Leary, M., Lapinska, K., Heerboth, S., Willbanks, A., & Sarkar, S. (2016). A Comparative Analysis of Genetic and Epigenetic Events of Breast and Ovarian Cancer Related to Tumorigenesis. International Journal of Molecular Sciences, 17(5), 759. https://doi.org/10.3390/ijms17050759