
Dendritic-Tumor Fusion Cell-Based Cancer Vaccines


Division of Gastroenterology and Hepatology, Department of Internal Medicine, The Jikei University School of Medicine, Kashiwa Hospital, 277-8567 Chiba, Japan
Int. J. Mol. Sci. 2016, 17(6), 828; https://doi.org/10.3390/ijms17060828
Submission received: 14 May 2016
/
Revised: 19 May 2016
/
Accepted: 23 May 2016
/
Published: 26 May 2016
(This article belongs to the Special Issue Cell Fusion in Cancer)
Abstract
:Dendritic cells (DCs) are potent antigen-presenting cells (APCs) that play a critical role in the induction of antitumor immunity. Therefore, various strategies have been developed to deliver tumor-associated antigens (TAAs) to DCs as cancer vaccines. The fusion of DCs and whole tumor cells to generate DC-tumor fusion cells (DC-tumor FCs) is an alternative strategy to treat cancer patients. The cell fusion method allows DCs to be exposed to the broad array of TAAs originally expressed by whole tumor cells. DCs then process TAAs endogenously and present them through major histocompatibility complex (MHC) class I and II pathways in the context of costimulatory molecules, resulting in simultaneous activation of both CD4+ and CD8+ T cells. DC-tumor FCs require optimized enhanced immunogenicity of both DCs and whole tumor cells. In this context, an effective fusion strategy also needs to produce immunogenic DC-tumor FCs. We discuss the potential ability of DC-tumor FCs and the recent progress in improving clinical outcomes by DC-tumor FC-based cancer vaccines.
1. Introduction
1.1. Dendritic Cell (DC)-Based Cancer Vaccines
T cell activation requires the processing of tumor-associated antigens (TAAs) into antigenic peptides, which are presented by antigen-presenting cells (APCs). Dendritic cells (DCs) are powerful APCs capable of inducing antitumor immune responses by linking the innate and adaptive immune systems [1,2]. Effective antigen processing and presentation by DCs are essential for the induction of antitumor immunity. There are three different pathways for antigen presentation by DCs. Exogenous antigens can be captured and processed into antigenic peptides by immature DCs, resulting in the cross-presentation of peptides on major histocompatibility complex (MHC) class I molecules [3,4]. Exogenous antigenic peptides can also be presented on MHC class II molecules on DCs [5]. In contrast, DCs can process endogenously synthesized antigens into antigenic peptides, which are presented on MHC class I molecules. DCs migrate to the draining lymph node where they present an antigen to CD4+ and CD8+ T cells, resulting in the induction of antigen-specific helper and cytotoxic T lymphocytes (CTLs), respectively. Moreover, interactions of DCs with innate and innate-like immune cells, such as natural killer (NK), invariant natural killer T (iNKT), and γδ T cells, generate crosstalk between DCs, resulting in efficient CTL induction [6,7,8]. Therefore, promising strategies to induce TAA-specific T cells have been developed using DCs [4]. In our laboratory, monocyte-derived immature DCs are generated by a single leukapheresis after culture in the presence of granulocyte macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-4, followed by activation with penicillin-killed and lyophilized preparations of a low-virulence strain (Su) of Streptococcus pyogenes (OK-432) and prostaglandin E2 (PGE2) [9]. A large number of mature DCs can then be cryopreserved in aliquots. However, the maturation of DCs with OK-432, PGE2, zoledronic acid, and tumor necrosis factor-α (TNF-α) may have more potential [10]. Thus far, mature DCs pulsed with specific TAA-derived peptides have been intensively investigated because they are simple and economical [11,12]. However, the major disadvantages of peptide-loading DC vaccines are related to the following factors: (1) the limited number of available immunogenic peptides specific for tumors [13]; (2) monoclonal peptide-specific CD8+ CTLs may not be sufficiently effective to treat cancer patients [14]; and (3) MHC class I molecules and certain TAAs are significantly downregulated in tumors during tumor progression [14]. Therefore, to induce polyclonal antigen-specific CTLs, DCs have also been loaded with TAAs in the form of tumor lysates [11], killed tumor cells [15], mRNA [16], and cDNA [17]. Moreover, an alternative strategy is the use of hybrid cells generated by the fusion of DCs and whole tumor cells (DC-tumor FCs), as first described by Gong et al. [18].
1.2. Fusions of Autologous DCs and Autologous Whole Tumor Cells
The fusion of DCs and whole tumor cells by chemical, physical, or biological means creates heterokaryons, which include DC-derived MHC class I, MHC class II, and costimulatory molecules as well as whole tumor-derived large repertories of TAAs [19,20,21,22] (Figure 1). We have used polyethylene glycol (PEG) to generate DC-tumor FCs [18]. PEG-generated DC-tumor FCs display tight contact between the DC and tumor cell, thus, efficiently integrating these two cell types [20,23]. In general, mature DCs and whole tumor cells are mixed at a 5–10:1 ratio in pre-warmed serum-free RPMI 1640 medium. The mixed cell pellet is gently resuspended with pre-warmed PEG solution for 3–5 min at room temperature followed by dilution with pre-warmed serum-free RPMI medium. The cell pellet obtained by gentle centrifugation at room temperature is washed and cultured in the presence of GM-CSF, IL-4, and OK-432. On day five or six of culture, the loosely-adherent cells are collected by gently pipetting up and down several times. During culture, the DCs and whole tumor cells are integrated into a single entity [19,23,24]. Whole tumor-whole tumor fusion cells, as well as unfused whole tumor cells, grow firmly attached to the culture plates, whereas DC-tumor FCs, unfused DCs, and DC-DC FCs adhere loosely to the culture plates. Short-term culture of PEG-treated cell preparations can promote DC-tumor fusion efficiency [23]. Although fusion efficiency is low immediately after the fusion process, one week of culture provides DC-tumor FCs sufficient time to integrate and display antigen in the context of MHC molecules [23]. However, prolonged culture should be avoided because unfused tumor cells can overgrow. Fusion efficiency also depends on cell conditions due to the sensitivity of cells to PEG treatment. PEG treatment is most suitable for fusions of living cells [20]. Moreover, DCs can also capture apoptotic whole tumor cells during culture. Therefore, special methods are not necessary to enrich DC-tumor FC preparations [20]. In clinical trials, DC-tumor FC preparations have been irradiated to prevent proliferation of unfused tumor cells. Thus, irradiated DC-tumor FC preparations are incapable of spreading in cancer patients [22].
In DC-tumor FCs, the cytoplasm of both DCs and whole tumor cells is integrated without nuclear fusion, as demonstrated by immunoelectron microscopy (Figure 1) [19,23]. These morphological features allow the retention of the functions of both original cell types, including co-expression of tumor-derived whole TAAs (both known and unidentified) and DC-derived MHC class I and II molecules [20,23]. In general, DC-tumor FCs process multiple antigenic peptides from whole tumor cells and load them onto MHC class I molecules in the endoplasmic reticulum. The antigenic peptide-MHC class I complexes are expressed on the DC-tumor FC surface and presented to CD8+ T cells. The endogenous pathway of direct antigen processing and presentation in DC-tumor FCs is preserved. DC-tumor FCs can also synthesize MHC class II-restricted antigenic peptides from whole tumor cells in the endoplasmic reticulum. DC-derived MHC class II molecules and tumor-derived antigenic peptides travel by separate routes and converge to form MHC class II-peptide complexes in DC-tumor FCs, where MHC class II-antigenic peptide complexes are expressed on the DC-tumor FC surface and presented to CD4+ T cells. Therefore, polyclonal antigen-specific CD4+ and CD8+ T cells are directly induced by DC-tumor FCs in the draining lymph node [20,25,26,27,28,29,30,31]. Moreover, antigens derived from DC-tumor FCs may also be cross-presented by host DCs, resulting in the induction of antitumor immunity [25]. Importantly, the effector and memory CD4+ T cells induced by DC-tumor FCs are crucial for the maintenance of long-term antitumor immunity [31]. A subset of primed MUC1-specific CD4+ T cells induced by DC/MUC1-positive tumor FCs possesses cytotoxicity against MHC class I- and MUC1-positive tumor cells [31,32]. Moreover, adoptive transfer of primed CD4+ T cells prevents lung metastasis in Rag2−/−mice that lack NKT, T, and B cells due to an impaired T cell receptor (TCR) rearrangement [31]. Therefore, CD4+ T cell activation by DC-tumor FCs through MHC class II interactions plays an essential role in the induction of effective antitumor immunity.
In summary, the DC-tumor fusion approach offers the following advantages for inducing antitumor immune responses (Table 1): (1) DC-tumor FCs present whole tumor-derived antigenic peptides, which avoids the need to identify antigenic peptides for individual patients; (2) a broad array of known and unidentified TAAs can be simultaneously presented on the surface of DC-tumor FCs, which increases the frequency of polyclonal antigen-specific CD4+ and CD8+ T cells, resulting in long-term efficient antitumor immunity; (3) numerous TAAs are presented in the context of co-stimulatory molecules, which prevents tolerance induction, resulting in efficient antitumor immune response; and (4) DC-tumor FCs migrate into draining lymph nodes and form clusters with CD4+ and CD8+ T cells in the T cell area of lymph nodes, such that DC-tumor FCs do not have to take up exogenous TAAs in order to activate CD4+ and CD8+ T cells.
1.3. Fusion of Autologous DCs and Allogeneic Whole Tumor Cells
The main disadvantage of the DC-tumor fusion approach is the limited availability of viable autologous tumor cells as a fusion partner due to the long culture time of primary tumor cells, as well as potential bacterial and fungal contamination, especially in gastrointestinal tumors [33]. Thus, DC-tumor FC-based vaccines may need to shift from autologous tumor cells to easily applicable tumor cells. To circumvent this disadvantage, allogeneic tumor cell lines have been used in place of autologous tumor cells to induce autologous tumor-specific antitumor immune responses (Figure 2) [33,34,35]. The scientific basis for this approach is that almost all allogeneic tumor cell lines share certain TAAs with autologous tumor cells. Antigenically well-defined allogeneic tumor cell lines sharing TAAs with autologous tumor cells can proliferate well in vitro under good manufacturing practice (GMP) standards, thus generating sufficient numbers of available tumor cells for a fusion partner. Moreover, allogeneic tumor cell lines are highly standardized in large-scale production vaccines [36]. For the DC-tumor fusion strategy, allogeneic tumor cell lines can theoretically process and present multiple TAAs through MHC class I and II molecules from autologous DCs. Our previous study indicated that fusions of autologous DCs and allogeneic tumor cell lines (e.g., colorectal cancer cells, breast cancer cells, or pancreatic cancer cells) present multiple TAAs and induce antigen-specific CTL responses restricted by autologous HLA [33,34,35]. The phenomenon of cross-priming against shared TAAs allows the use of allogeneic tumor cell lines to present antigenic peptides to autologous DCs in fusions of autologous DCs and allogeneic tumor cells. Therefore, it is not necessary to match HLA types between cancer patients and allogeneic tumor cells used for generating fusions [33,34]. In addition, allogeneic MHC molecules from tumor cell lines may induce allogeneic responses, promoting effective antitumor immunity [37]. Thus far, clinical trials have not reported using DC-tumor FCs generated with allogeneic tumor cell lines.
1.4. Fusion of Allogeneic DCs and Autologous Whole Tumor Cells
Cancer patient-derived DCs are sometimes defective as APCs due to tumors, tumor microenvironments, and cancer therapies [38]. Therefore, the use of DCs from healthy donors as a source of allogeneic DCs to generate allogeneic DC-tumor FC vaccines has been investigated (Figure 3) [39]. Advantages of this strategy are that DCs generated from healthy donors have functional APCs and are readily available in sufficient quantities. Allogeneic DC-tumor FCs express DC-derived allogeneic HLA class II molecules for direct stimulation of alloreactive CD4+ T cells. This allogeneic response may contribute to enhanced and maintained antigen-specific CTL responses by cytokines from alloreactive CD4+ T cells [37]. We have generated allogeneic DC-tumor FCs in murine [40,41,42], preclinical [43], and clinical [44] studies, and we have shown equal or less potential of allogeneic DC-tumor FCs with autologous DC-tumor FCs. If an allogenically-healthy donor and cancer patient do not share any HLA molecules, the patient’s MHC-restricted presentation of antigenic peptides by allogeneic DC-tumor FCs is not possible. If the HLA typing is shared between the healthy donor and cancer patient, the antigen-specific CTL induction changes dramatically [37]. Where there is some sharing of MHC class I molecules, the alloreactive CD4+ T cell response provides potent T cell help for the generation of CD8+ CTL responses to tumor peptides presented by the shared HLA class I molecules. Antigen-specific CD4+ and CD8+ T cell responses against autologous tumor cells induced by allogeneic DC-tumor FCs are dependent on HLA type. Therefore, fusions of autologous DCs and autologous tumor cells may be more effective in targeting tumor cells because they can stimulate antigen-specific CD4+ T cells.
1.5. Fusion of Allogeneic DCs and Allogeneic Whole Tumor Cells
Allogeneic DC lines and allogeneic tumor cell lines may be used instead of autologous cells. Cell lines are well characterized and can be well propagated in vitro under GMP standards. Therefore, unlimited amounts of allogeneic DC-allogeneic tumor FCs can be readily available (Figure 4). DC-tumor FC vaccines with fully allogeneic components have been demonstrated to induce clinical responses [45]. Moreover, we have shown that fusion cells generated with an allogeneic DC line and allogeneic tumor cell line can induce antigen-specific CTL responses in vitro [46]. These findings introduce the possibility of using defined allogeneic DCs and allogeneic tumor lines to induce antigen-specific CTLs for adoptive immunotherapy. Although DC-tumor FCs generated with fully syngeneic components may be most effective for antigen-specific CTL induction, semi-allogeneic and fully-allogeneic components may also have potential in the field of adoptive immunotherapy [47].
1.6. Fusion of Activated Autologous DCs and Autologous Whole Tumor Cells
Although DC-tumor FC vaccines are safe and have induced efficient antitumor immune responses in early clinical trials, a limited number of efficient clinical responses has been reported in glioblastoma, myeloma, melanoma, gastric cancer, breast cancer, or renal cell cancer patients [21,22,39,48]. To maximize the induction of antitumor immunity by vaccination with DC-tumor FCs, many types of adjuvants have accompanied DC-tumor FCs in animal studies. For example, adjuvants, including IL-2 [49], IL-12 [50,51,52], IL-18 [52,53], oligodeoxynucleotides containing a CpG motif (CpG ODN) [53,54], 1-MT (an indoleamine-pyrrole 2,3-dioxygenase inhibitor) [55], or polyriboinosinic polyribocytidylic acid [Poly(I:C)]/IL-10 siRNA, have been used [56]. Therefore, in clinical trials, DC-tumor FCs must be co-administered with adjuvants to induce efficient clinical responses in advanced cancer patients. Despite the small quantity of FCs, vaccination using DC-tumor FCs with IL-12 has demonstrated better therapeutic responses compared with DC-tumor FCs alone [57,58]. Co-administration of IL-12 with DC-tumor FCs may promote antitumor T helper type 1 (Th1) polarization, resulting in the activation of NK cells and antigen-specific CTLs [59].
Modification of DC-tumor FCs is also required to treat advanced cancer patients. Interestingly, both DCs and whole tumor cells can be independently immunomodulated while maintaining their individual characteristics even after fusion. This characteristic of DC-tumor FCs is advantageous compared to the strategy of whole tumor cell lysate loading of DCs. Recognition of microbial stimuli by Toll-like receptors (TLRs) expressed on DCs is effective for generating immunogenic DCs, leading to the production of Th1 cytokines, such as IL-12, and co-stimulatory molecules, such as CD80 and CD86, in DCs [60,61]. Full activation of DCs requires receptor signaling by combined TLR agonists [62]. The combination of TLR2 and TLR4 agonists has been shown to promote the immunogenicity of fusions, as demonstrated by the following results: (1) increased fusion efficiency; (2) upregulation of MHC class II molecules, heat shock proteins (HSPs), and CD86 expression in DC-tumor FCs; (3) increased IL-12 production from DC-tumor FCs; (4) activation of antigen-specific polyclonal CD4+ and CD8+ T cells producing high levels of interferon-γ (IFN-γ); and (5) induction of augmented antitumor immunity [39,63,64]. Therefore, the combination of TLR2 and TLR4 agonists for DC activation is advantageous and has potential application in immunogenic fusions for cancer vaccines.
1.7. Fusion of Autologous DCs and Immunogenic Autologous Whole Tumor Cells
Whole tumor cells are also required for the generation of immunogenic DC-tumor FCs. Indeed, many kinds of tumor cells produce immunosuppressive molecules, such as transforming growth factor β (TGF-β), IL-10, and vascular endothelial growth factor (VEGF). In particular, TGF-β1 is one of the major cytokines that impair DC function [65] and generate regulatory T cells (Tregs) [66], which play an important role in the inactivation of antitumor immunity [67]. TGF-β1 produced from whole tumor cells decreases antitumor immunity by DC-tumor FCs even when TLR2 and TLR4 are co-administered [64]. Therefore, several strategies have been developed to improve poor immunogenicity in tumor cells. In one fusion strategy, TGF-β production from whole tumor cells is blocked by a soluble TGF-β receptor expressing DC-tumor FCs, resulting in reduced Treg generation and augmented antitumor immunity [68]. As compared to conventional DC-tumor FCs, we have previously demonstrated that fusions generated with DCs and heat- [63] or ethanol-treated [69] tumor cells upregulate multiple HSPs, MHC class I molecules, MHC class II molecules, TAAs, CD80, CD86, and IL-12. Therefore, the fusion of DCs and immunogenic whole tumor cells activates antigen-specific CD4+ and CD8+ T cells that produce high levels of IFN-γ. Therefore, immunogenic whole tumor cells induced by heat or ethanol treatment enhance the immunogenicity of DC-tumor FCs as cancer vaccines. Interestingly, TGF-β1 production from tumor cells is also easily blocked by exposure to ethanol without downregulating MHC class I molecules and TAAs [69]. Moreover, inducing immunogenic tumor cell death by ethanol treatment ectopically induces calreticulin and high-mobility group box 1 (HMGB1), which activate DCs [70,71]. Immunogenic tumor cells might be used as whole tumor cell-based cancer vaccines. In clinical trials, allogeneic tumor cell lines have been transduced with GM-CSF to generate immunogenic whole tumor cells to be used for cancer vaccines (GVAX), leading to augmented antitumor immunity in early clinical trials [72]. One disadvantage of using heat- or ethanol-treated whole allogeneic tumor cells is the necessary dose response test to evaluate the optimal conditions for generating immunogenic whole tumor cells. The fusion strategy generated with activated DCs by TLRs and immunogenic whole tumor cells synergistically yields efficient TAA processing and presentation by DC-tumor FCs, with upregulated expression of MHC class I molecules, MHC class II molecules, co-stimulatory molecules, TAAs, and IL-12 [63,73,74]. However, it remains unclear which specific agents, including cytotoxic chemotherapeutic agents, or irradiation processes are best suited to generate immunogenic tumor cells. Thus, optimal strategies to maximize the clinical outcomes of DC-tumor FCs-based cancer vaccines must be identified.
1.8. Fusion of DCs and Cancer Stem Cells
It is well accepted that cancer stem cells (CSCs) are resistant to standard therapies, such as chemotherapy and irradiation [75,76]. Therefore, small populations of chemoresistant CSCs are responsible for lethal events, including tumor relapse and growth, due to therapeutic failure [75,76]. Importantly, chemoresistant CSCs preferentially express stem cell markers, including OCT3/4, ABCG2, nestin, SOX2, Bmi-1, Notch-1, CD44, CD133, and CD177 [75,77]. Moreover, CSCs overexpress survivin, MUC1, hTERT, HER2, CERP55, COA-1, and WT1 [77,78]. Thus, CSCs remain attractive targets for cancer vaccines, and the success of cancer vaccines may at least partly depend on the efficient induction of anti-CSC immunity. We have reported that fusions of DCs and CD44+ tumor cells with CSC characteristics (e.g., spheroid formation in culture, self-renewal, and an ability to be engrafted in immunocompromised mice) can endogenously process and present multiple CSC-specific antigenic peptides on MHC class I and II molecules, resulting in the induction of efficient CSC-specific CTL responses [76]. Moreover, another study demonstrated that fusions of DCs and CD133+ tumor cells could induce antitumor immunity similar to that of DC and CD133-tumor cell fusions [79]. Fusion cells generated with DCs and CSCs (DC-CSC-FCs) can process and present whole CSC antigens, thus avoiding the need for identification of CSC-specific antigens. Therefore, DC-CSC-FC vaccines may be an alternative approach to induce efficient anti-CSC immunity. Interestingly, MUC1 expression is upregulated in chemoresistant CSCs that are efficiently lysed by MUC1-specific CTLs in mice [76,80]. Therefore, DC-CSC-FCs should be combined with conventional chemotherapy.
1.9. Chaperone-Peptide Complexes from DC-Tumor Fusion Cells (FCs)
Cancer vaccines based on HSP-peptide complexes are promising because molecular chaperones, such as HSP70 and HSP90, have been shown to form complexes with a wide panel of peptides, including antigenic peptides [81,82]. HSP-peptide complexes can be taken up by DCs through specific receptors, facilitating DC activation and promoting antigen processing and presentation by MHC class I and II molecules on the DC surface [83]. We and other groups have attempted to prepare cancer vaccines based on HSP-peptide complexes derived from DC-tumor FCs [84,85,86,87]. We have demonstrated that HSP70-peptide complexes (HSP70.PC) extracted from DC-tumor FCs (HSP70.PC-FCs) have a superior ability to activate DCs and induce antigen-specific CTLs compared to HSP70.PC from whole tumor cells (HSP70.PC-T) in mice [84] and in preclinical studies [85]. Therefore, the DC-tumor FC strategy is useful in the preparation of HSP70.PC-based cancer vaccines in patients. This alternative approach relies on the efficient antigen processing and presentation machinery of DC-tumor FCs [85].
1.10. DC-Tumor FC Combination Therapy
As standard cancer therapies, such as chemotherapy, radiotherapy, and targeted therapies, interfere with DNA synthesis of tumor cells and peripheral lymphocytes, the therapy may blunt antitumor immune responses. However, increasing evidence suggests that some chemotherapeutic agents have the potential to augment cancer vaccines. Chemotherapy agents or irradiation can promote intrinsic immunogenicity of whole tumor cells in multiple ways. For example, sublethal therapies undergo immunogenic modulation and demonstrate upregulation of various danger signals, such as HMGB1, HSP70/90, adenosine triphosphate (ATP), and calreticulin (CRT) [70,88,89,90]. CRT is a Ca2+-binding chaperone that is found in different apoptotic stages and leads to increased cross-priming of antitumor T cell immunity without Treg induction [70,88,89,91,92,93]. Moreover, HMGB1 and HSP70/90 from immunogenic tumor cells interact with TLR4 on DCs, which activates the antigen processing and presentation machinery in DCs [71,94,95]. Some therapies also upregulate TAAs, such as WT1, CEA, MUC1, and Her2, as well as MHC class I molecules [96,97,98,99]. Finally, a positive interaction of DC-tumor FC-based cancer vaccines with chemotherapy or radiotherapy may generate a new era for cancer immunotherapy. Moreover, chemotherapy- or radiotherapy-induced immunogenic tumor modulation is a better candidate as a fusion partner than conventional DC-tumor FCs.
1.11. Future Cancer Regimens Using DC-Tumor FCs
Effective and selective targeted therapies with little toxicity are urgently needed for patients with advanced cancer. Treatment of cancer patients with DC-tumor FCs alone has limitations due to the immunosuppressive mechanism. DC-tumor FCs can induce antigen-specific CTLs and Tregs [100]. It is well known that some chemotherapeutic agents, such as cyclophosphamide and gemcitabine, can activate antitumor immunity by depleting Tregs and myeloid-derived suppresser cells (MDSCs) [101,102], resulting in efficient clinical outcomes. Recent reports have also indicated that vaccination induces programmed death 1 (PD1) expression in activated CTLs [103]. The PD1 ligand, PD-L1, in tumor cells is upregulated by IFN-γ produced by activated CTLs. As a result, PD1-PD-L1 signaling is associated with impaired CTL function. A preclinical study has shown that an anti-PD1 antibody decreases Tregs and enhances CTL activity [104]. Moreover, inactivation of CD4+CD25+Foxp3+ Tregs by an anti-CD25 antibody following DC-tumor FC vaccination significantly improves antitumor immunity in a murine model [105]. Therefore, a therapeutic regimen combining DC-tumor FCs, chemotherapy, Treg depletion, and antibody blockade of PD1-PD-L1 signaling may have potential in advanced cancer patients [21,106]. Such multiple immune checkpoint blockades to facilitate CTL induction without T cell anergy in the presence of DC-tumor FC-augmented CTLs may be the most efficient treatment strategy. It is important to understand which combinations of immunoinhibitory molecules and which patients are suitable for the therapies [107].
2. Conclusions
We developed DC-tumor FCs strategies to induce efficient antitumor immune responses mediated by antigen-specific CD4+ and CD8+ T cells and to break T cell tolerance to TAAs. Although DC-tumor FCs strategy has numerous advantages, the clinical responses to DC-tumor FCs are not as vigorous as in the animal tumor models. Therefore, DC-tumor FCs require optimized enhanced immunogenicity of both DCs and whole tumor cells to maximize their immunogenicity. Moreover, the combination of DC-tumor FCs with standard therapies such chemotherapy and irradiation may provide robust clinical benefits. Specially, an immune checkpoint blockade combined with DC-tumor FCs may be effective in treating patients with advanced cancer. It is also essential to develop strategies targeting immunosuppressive microenvironment of tumors.
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research (C) from the Ministry of Education, Cultures, Sports, Science, and Technology, Tokyo, Japan.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ATP | Adenosine triphosphate |
| APCs | Antigen presenting cells |
| CRT | Calreticulin |
| CSCs | Cancer stem cells |
| CTLs | Cytotoxic T lymphocytes |
| DCs | Dendritic cells |
| DC-tumor FCs | Fusions of DCs and whole tumor cells |
| GMP | Good manufacturing practice |
| GMP | Granulocyte macrophage colony-stimulating factor |
| HSPs | Heat shock proteins |
| HSP70.PC | HSP70-peptide complexes |
| HSP70.PC-FCs | HSP70.PC from DC-tumor FCs |
| HSP70.PC-T | HSP70.PC from whole tumor cells |
| IFN-γ | Interferon-γ |
| IL | Interleukin |
| iNKT | Invariant natural killer T |
| MHC | Major histocompatibility complex |
| MDSCs | Myeloid derived suppresser cells |
| NK | Natural killer |
| Poly(I:C) | Polyriboinosinic polyribocytidylic acid |
| PEG | Polyethylene glycol |
| PD1 | Programmed death 1 |
| PD-L1 | Programmed death ligand 1 |
| PGE2 | Prostaglandin E2 |
| Tregs | Regulatory T cells |
| Th1 | T helper type 1 |
| TLR | Toll-like receptor |
| TGF-β | Transforming growth factor β |
| TAAs | Tumor-associated antigens |
| TNF-α | Tumor necrosis factor-α |
| VEGF | Vascular endothelial growth factor |
References
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Swanson, J. The endocytic activity of dendritic cells. J. Exp. Med. 1995, 182, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Bloy, N.; Pol, J.; Aranda, F.; Eggermont, A.; Cremer, I.; Fridman, W.H.; Fučíková, J.; Galon, J.; Tartour, E.; Spisek, R.; et al. Trial watch: Dendritic cell-based anticancer therapy. Oncoimmunology 2014, 3, e963424. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, J.; Bourgeois-Daigneault, M.; Lapointe, R. Targeting the MHC class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology 2012, 1, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, J.J.; Wimmers, F.; Hato, S.V.; de Vries, I.J.; Sköld, A.E. Dendritic cell cross talk with innate and innate-like effector cells in antitumor immunity: Implications for DC vaccination. Crit. Rev. Immunol. 2014, 34, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; King, S.; Allgeier, T.; Braumüller, H.; Lüking, C.; Mysliwietz, J.; Kriegeskorte, A.; Busch, D.H.; Röcken, M.; Mocikat, R. DC-NK cell cross talk as a novel CD4+ T-cell-independent pathway for antitumor CTL induction. Blood 2005, 106, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Wehner, R.; Dietze, K.; Bachmann, M.; Schmitz, M. The bidirectional crosstalk between human dendritic cells and natural killer cells. J. Innate Immun. 2011, 3, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Takakura, K.; Mori, M.; Yoshizaki, S.; Tsukinaga, S.; Odahara, S.; Koyama, S.; Imazu, H.; et al. Treatment with chemotherapy and dendritic cells pulsed with multiple Wilms’ tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin. Cancer Res. 2014, 20, 4228–4239. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Nagaoka, K.; Takahara, M.; Yang, X.Y.; Liu, C.X.; Guo, H.; Roy Choudhury, K.; Hobeika, A.; Hartman, Z.; Morse, M.A.; et al. Precision cancer immunotherapy: Optimizing dendritic cell-based strategies to induce tumor antigen-specific T-cell responses against individual patient tumors. J. Immunother. 2015, 38, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Bloy, N.; Buqué, A.; Eggermont, A.; Cremer, I.; Sautès-Fridman, C.; Galon, J.; Tartour, E.; Zitvogel, L.; Kroemer, G.; et al. Trial watch: Peptide-based anticancer vaccines. Oncoimmunology 2015, 4, e974411. [Google Scholar] [CrossRef] [PubMed]
- Kanodia, S.; Kast, W.M. Peptide-based vaccines for cancer: Realizing their potential. Expert Rev. Vaccines 2008, 7, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. Immunotherapy: Past, present and future. Nat. Med. 2003, 9, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Ueno, H.; Connolly, J.; Kerneis-Norvell, F.; Blanck, J.P.; Johnston, D.A.; Fay, J.; Banchereau, J. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and Mart-1 specific CD8+ T-cell immunity. J. Immunother. 2006, 29, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, E.; Nair, S.K.; Lyerly, H.K. Immunotherapy of cancer with dendritic-cell-based vaccines. Cancer Immunol. Immunother. 1998, 46, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Bubeník, J. Genetically engineered dendritic cell-based cancer vaccines (Review). Int. J. Oncol. 2001, 18, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chen, L.; Chen, D.; Kashiwaba, M.; Manome, Y.; Tanaka, T.; Kufe, D. Induction of antigen-specific antitumor immunity with adenovirus-transduced dendritic cells. Gene Ther. 1997, 4, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Ohana, M.; Liu, C.; Nikrui, N.; Durfee, J.; Lerner, A.; Gong, J. Dendritic cells fused with human cancer cells: Morphology, antigen expression, and T cell stimulation. Clin. Immunol. 2004, 113, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Koido, S.; Calderwood, S.K. Cell fusion: From hybridoma to dendritic cell-based vaccine. Expert Rev. Vaccines 2008, 7, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, M.; Takakura, K.; Ohkusa, T.; Koido, S. The impact of dendritic cell-tumor fusion cells on cancer vaccines—Past progress and future strategies. Immunotherapy 2015, 7, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Takakura, K.; Kajihara, M.; Ito, Z.; Ohkusa, T.; Gong, J.; Koido, S. Dendritic-tumor fusion cells in cancer immunotherapy. Discov. Med. 2015, 19, 169–174. [Google Scholar] [PubMed]
- Koido, S.; Gong, J. Characterization of structure and direct antigen presentation by dendritic/tumor-fused cells as cancer vaccines. Anticancer Res. 2013, 33, 347–354. [Google Scholar] [PubMed]
- Koido, S.; Gong, J. Cell fusion between dendritic cells and whole tumor cells. Methods Mol. Biol. 2015, 1313, 185–191. [Google Scholar] [PubMed]
- Koido, S.; Tanaka, Y.; Chen, D.; Kufe, D.; Gong, J. The kinetics of in vivo priming of CD4 and CD8 T cells by dendritic/tumor fusion cells in MUC1-transgenic mice. J. Immunol. 2002, 168, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Nikrui, N.; Ohana, M.; Xia, J.; Tanaka, Y.; Liu, C.; Durfee, J.K.; Lerner, A.; Gong, J. Assessment of fusion cells from patient-derived ovarian carcinoma cells and dendritic cells as a vaccine for clinical use. Gynecol. Oncol. 2005, 99, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Torii, A.; Homma, S.; Toyama, Y.; Kawahara, H.; Ogawa, M.; Watanabe, M.; Yanaga, K.; Fujise, K.; et al. Induction of antigen-specific CD4- and CD8-mediated T-cell responses by fusions of autologous dendritic cells and metastatic colorectal cancer cells. Int. J. Cancer 2005, 117, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Fujise, K.; Gong, J.; Tajiri, H. Dendritic/tumor fusion cell-based vaccination against cancer. Arch. Immunol. Ther. Exp. 2007, 55, 281–287. [Google Scholar] [CrossRef]
- Koido, S.; Homma, S.; Hara, E.; Namiki, Y.; Takahara, A.; Komita, H.; Nagasaki, E.; Ito, M.; Ohkusa, T.; Gong, J.; et al. Regulation of tumor immunity by tumor/dendritic cell fusions. Clin. Dev. Immunol. 2010, 2010, 516768. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Uchiyama, K.; Kajihara, M.; Arihiro, S.; Imazu, H.; Arakawa, H.; et al. Fusions between dendritic cells and whole tumor cells as anticancer vaccines. Oncoimmunology 2013, 2, e24437. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Enomoto, Y.; Apostolopoulos, V.; Gong, J. Tumor regression by CD4 T-cells primed with dendritic/tumor fusion cell vaccines. Anticancer Res. 2014, 34, 3917–3924. [Google Scholar] [PubMed]
- Tanaka, Y.; Koido, S.; Ohana, M.; Liu, C.; Gong, J. Induction of impaired antitumor immunity by fusion of MHC class II-deficient dendritic cells with tumor cells. J. Immunol. 2005, 174, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Torii, A.; Toyama, Y.; Kawahara, H.; Watanabe, M.; Yanaga, K.; Fujise, K.; Tajiri, H.; et al. Dendritic cells fused with allogeneic colorectal cancer cell line present multiple colorectal cancer-specific antigens and induce antitumor immunity against autologous tumor cells. Clin. Cancer Res. 2005, 11, 7891–7900. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Tanaka, Y.; Tajiri, H.; Gong, J. Generation and functional assessment of antigen-specific T cells stimulated by fusions of dendritic cells and allogeneic breast cancer cells. Vaccine 2007, 25, 2610–2619. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Namiki, Y.; Komita, H.; Takahara, A.; Nagasaki, E.; Ito, M.; Sagawa, Y.; Mitsunaga, M.; et al. Dendritic/pancreatic carcinoma fusions for clinical use: Comparative functional analysis of healthy- versus patient-derived fusions. Clin. Immunol. 2010, 135, 384–400. [Google Scholar] [CrossRef] [PubMed]
- de Gruijl, T.D.; van den Eertwegh, A.J.; Pinedo, H.M.; Scheper, R.J. Whole-cell cancer vaccination: From autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol. Immunother. 2008, 57, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Fabre, J.W. The allogeneic response and tumor immunity. Nat. Med. 2001, 7, 649–652. [Google Scholar] [CrossRef] [PubMed]
- atthaporn, S.; Robins, A.; Vassanasiri, W.; El-Sheemy, M.; Jibril, J.A.; Clark, D.; Valerio, D.; Eremin, O. Dendritic cells are dysfunctional in patients with operable breast cancer. Cancer Immunol. Immunother. 2004, 53, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Ohkusa, T.; Gong, J.; Tajiri, H. Cancer immunotherapy by fusions of dendritic cells and tumor cells. Immunotherapy 2009, 1, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Koido, S.; Chen, D.; Gendler, S.J.; Kufe, D.; Gong, J. Vaccination with allogeneic dendritic cells fused to carcinoma cells induces antitumor immunity in MUC1 transgenic mice. Clin. Immunol. 2001, 101, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Kamigaki, T.; Kawasaki, K.; Nakamura, T.; Yamamoto, M.; Kanemitsu, K.; Takase, S.; Kuroda, D.; Kim, Y.; Ajiki, T.; et al. Superior anti-tumor protection and therapeutic efficacy of vaccination with allogeneic and semiallogeneic dendritic cell/tumor cell fusion hybrids for murine colon adenocarcinoma. Cancer Immunol. Immunother. 2007, 56, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.W.; Cowled, C.J.; Darling, D.; Guinn, B.A.; Farzaneh, F.; Noble, A.; Galea-Lauri, J. Semi-allogeneic dendritic cells can induce antigen-specific T-cell activation, which is not enhanced by concurrent alloreactivity. Cancer Immunol. Immunother. 2007, 56, 1861–1873. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Nikrui, N.; Chen, D.; Koido, S.; Wu, Z.; Tanaka, Y.; Cannistra, S.; Avigan, D.; Kufe, D. Fusions of human ovarian carcinoma cells with autologous or allogeneic dendritic cells induce antitumor immunity. J. Immunol. 2000, 165, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Avigan, D.E.; Vasir, B.; George, D.J.; Oh, W.K.; Atkins, M.B.; McDermott, D.F.; Kantoff, P.W.; Figlin, R.A.; Vasconcelles, M.J.; Xu, Y.; et al. Phase I/II study of vaccination with electrofused allogeneic dendritic cells/autologous tumor-derived cells in patients with stage IV renal cell carcinoma. J. Immunother. 2007, 30, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Märten, A.; Renoth, S.; Heinicke, T.; Albers, P.; Pauli, A.; Mey, U.; Caspari, R.; Flieger, D.; Hanfland, P.; von Ruecker, A.; et al. Allogeneic dendritic cells fused with tumor cells: Preclinical results and outcome of a clinical phase I/II trial in patients with metastatic renal cell carcinoma. Hum. Gene Ther. 2003, 14, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Kan, S.; Takakura, K.; Namiki, Y.; Kobayashi, H.; Ito, Z.; Uchiyama, K.; Kajihara, M.; Arihiro, S.; et al. Induction of antigen-specific cytotoxic T lymphocytes by fusion cells generated from allogeneic plasmacytoid dendritic and tumor cells. Int. J. Oncol. 2014, 45, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Siders, W.M.; Garron, C.; Shields, J.; Kaplan, J.M. Induction of antitumor immunity by semi-allogeneic and fully allogeneic electrofusion products of tumor cells and dendritic cells. Clin. Transl. Sci. 2009, 2, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Namiki, Y.; Ohkusa, T.; Gong, J.; Tajiri, H. Cancer vaccine by fusions of dendritic and cancer cells. Clin. Dev. Immunol. 2009, 2009, 657369. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, F.; Iinuma, H.; Okinaga, K. Dendritic cell vaccine therapy by immunization with fusion cells of interleukin-2 gene-transduced, spleen-derived dendritic cells and tumour cells. Scand. J. Immunol. 2004, 59, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Koido, S.; Chen, D.; Tanaka, Y.; Huang, L.; Avigan, D.; Anderson, K.; Ohno, T.; Kufe, D. Immunization against murine multiple myeloma with fusions of dendritic and plasmacytoma cells is potentiated by interleukin 12. Blood 2002, 12, 2512–2517. [Google Scholar] [CrossRef]
- Iinuma, T.; Homma, S.; Noda, T.; Kufe, D.; Ohno, T.; Toda, G. Prevention of gastrointestinal tumors based on adenomatous polyposis coli gene mutation by dendritic cell vaccine. J. Clin. Investig. 2004, 113, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, H.; Okinaga, K.; Fukushima, R.; Inaba, T.; Iwasaki, K.; Okinaga, A.; Takahashi, I.; Kaneko, M. Superior protective and therapeutic effects of IL-12 and IL-18 gene-transduced dendritic neuroblastoma fusion cells on liver metastasis of murine neuroblastoma. J. Immunol. 2006, 176, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Vasir, B.; Wu, Z.; Crawford, K.; Rosenblatt, J.; Zarwan, C.; Bissonnette, A.; Kufe, D.; Avigan, D. Fusions of dendritic cells with breast carcinoma stimulate the expansion of regulatory T cells while concomitant exposure to IL-12, CpG oligodeoxynucleotides, and anti-CD3/CD28 promotes the expansion of activated tumor reactive cells. J. Immunol. 2008, 181, 808–821. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, K.; Yamamoto, S.; Otsuru, S.; Nakai, S.; Tamai, K.; Morishita, R.; Ogihara, T.; Kaneda, Y. Enhanced tumor-specific long-term immunity of hemagglutinating [correction of hemaggluttinating] virus of Japan-mediated dendritic cell-tumor fused cell vaccination by coadministration with CpG oligodeoxynucleotides. J. Immunol. 2004, 173, 4297–4307. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Cai, S.; Liu, P.; Zeng, J.; He, Y.; Wu, X.; Du, J. Enhancement of dendritic cell-tumor fusion vaccine potency by indoleamine-pyrrole 2,3-dioxygenase inhibitor, 1-MT. J. Cancer Res. Clin. Oncol. 2008, 134, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Kikuchi, T.; Irie, M.; Yamamoto, Y.; Arai, T.; Tanaka, T.; Joki, T.; Abe, T. Cotransfection of poly(I: C) and siRNA of IL-10 into fusions of dendritic and glioma cells enhances antitumor T helper type 1 induction in patients with glioma. J. Immunother. 2011, 34, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Akasaki, Y.; Abe, T.; Fukuda, T.; Saotome, H.; Ryan, J.L.; Kufe, D.W.; Ohno, T. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J. Immunother. 2004, 27, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Akasaki, Y.; Irie, M.; Homma, S.; Abe, T.; Ohno, T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol. Immunother. 2001, 50, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; Pearce, E.J. Metabolic control of dendritic cell activation and function: Recent advances and clinical implications. Front. Immunol. 2014, 5, 203. [Google Scholar] [CrossRef] [PubMed]
- Warger, T.; Osterloh, P.; Rechtsteiner, G.; Fassbender, M.; Heib, V.; Schmid, B.; Schmitt, E.; Schild, H.; Radsak, M.P. Synergistic activation of dendritic cells by combined toll-like receptor ligation induces superior CTL responses in vivo. Blood 2006, 108, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Napolitani, G.; Rinaldi, A.; Bertoni, F.; Sallusto, F.; Lanzavecchia, A. Selected toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 2005, 6, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Mitsunaga, M.; Takahara, A.; Nagasaki, E.; Kawahara, H.; Watanabe, M.; Toyama, Y.; Yanagisawa, S.; et al. Synergistic induction of antigen-specific CTL by fusions of TLR-stimulated dendritic cells and heat-stressed tumor cells. J. Immunol. 2007, 179, 4874–4883. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Takahara, A.; Odahara, S.; Tsukinaga, S.; Yukawa, T.; Mitobe, J.; et al. Combined TLR2/4-activated dendritic/tumor cell fusions induce augmented cytotoxic T lymphocytes. PLoS ONE 2013, 8, e59280. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Gilmour, J.W.; Imami, N.; Amjadi, P.; Henderson, D.C.; Allen-Mersh, T.G. Increased serum transforming growth factor-β1 in human colorectal cancer correlates with reduced circulating dendritic cells and increased colonic langerhans cell infiltration. Clin. Exp. Immunol. 2003, 134, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H. Suppression of antitumor immunity by IL-10 and TGF-β-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Inge, T.H.; Hoover, S.K.; Susskind, B.M.; Barrett, S.K.; Bear, H.D. Inhibition of tumor-specific cytotoxic T-lymphocyte responses by transforming growth factor β 1. Cancer Res. 1992, 52, 1386–1392. [Google Scholar] [PubMed]
- Zhang, M.; Berndt, B.E.; Chen, J.J.; Kao, J.Y. Expression of a soluble TGF-β receptor by tumor cells enhances dendritic cell/tumor fusion vaccine efficacy. J. Immunol. 2008, 181, 3690–3697. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Kan, S.; Takakura, K.; Kajihara, M.; Uchiyama, K.; Hara, E.; Ohkusa, T.; et al. Improved immunogenicity of fusions between ethanol-treated cancer cells and dendritic cells exposed to dual TLR stimulation. Oncoimmunology 2013, 2, e25375. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Copier, J.; Dalgleish, A. Whole-cell vaccines: A failure or a success waiting to happen? Curr. Opin. Mol. Ther. 2010, 12, 14–20. [Google Scholar] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Takahara, A.; Odahara, S.; Tsukinaga, S.; Yukawa, T.; Mitobe, J.; et al. Augmentation of antitumor immunity by fusions of ethanol-treated tumor cells and dendritic cells stimulated via Dual TLRs through TGF-β1 blockade and IL-12p70 production. PLoS ONE 2013, 8, e63498. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Uchiyama, K.; Kajihara, M.; Arihiro, S.; Imazu, H.; Arakawa, H.; et al. Strategies to improve the immunogenicity of anticancer vaccines based on dendritic cell/malignant cell fusions. Oncoimmunology 2013, 2, e25994. [Google Scholar] [CrossRef]
- Hirohashi, Y.; Torigoe, T.; Inoda, S.; Takahashi, A.; Morita, R.; Nishizawa, S.; Tamura, Y.; Suzuki, H.; Toyota, M.; Sato, N. Immune response against tumor antigens expressed on human cancer stem-like cells/tumor-initiating cells. Immunotherapy 2010, 2, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Li, F.; Luo, B.; Wang, X.H.; Sun, H.C.; Liu, S.; Cui, Y.Q.; Xu, X.X. A side population of cells from a human pancreatic carcinoma cell line harbors cancer stem cell characteristics. Neoplasma 2009, 56, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Hirohashi, Y.; Torigoe, T.; Tsukahara, T.; Kanaseki, T.; Kochin, V.; Sato, N. Immune responses to human cancer stem-like cells/cancer-initiating cells. Cancer Sci. 2016, 107, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A National Cancer Institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Tian, G.; Li, P.; Chen, Q.; Zhang, R.; Ke, Y.Q.; Xiao, Z.C.; Jiang, X.D. Anti-glioma response of autologous T cells stimulated by autologous dendritic cells electrofused with CD133+ or CD133− glioma cells. J. Neuroimmunol. 2012, 242, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, K.; Shen, H.; Finn, O.J. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 2008, 68, 2419–2426. [Google Scholar] [CrossRef] [PubMed]
- Murshid, A.; Gong, J.; Calderwood, S.K. Purification, preparation, and use of chaperone-peptide complexes for tumor immunotherapy. Methods Mol. Biol. 2013, 960, 209–217. [Google Scholar] [PubMed]
- Murshid, A.; Gong, J.; Calderwood, S.K. Hsp90-peptide complexes stimulate antigen presentation through the class II pathway after binding scavenger receptor SREC-I. Immunobiology 2014, 219, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, C.; Welch, W.J. Role of the major heat shock proteins as molecular chaperones. Annu. Rev. Cell Biol. 1993, 9, 601–634. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Bharti, A.; Khaleque, A.A.; Song, B.; Liu, C.; Apostolopoulos, V.; Xing, P.X.; Calderwood, S.K.; Gong, J. Enhanced immunogenicity of heat shock protein 70 peptide complexes from dendritic cell-tumor fusion cells. J. Immunol. 2006, 177, 5946–5955. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhang, Y.; Durfee, J.; Weng, D.; Liu, C.; Koido, S.; Song, B.; Apostolopoulos, V.; Calderwood, S.K. A heat shock protein 70-based vaccine with enhanced immunogenicity for clinical use. J. Immunol. 2010, 184, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, W.; Wang, Y.; Chen, J.; Liu, Y.; Zhang, Y. Enhanced antitumor immunity of nanoliposome-encapsulated heat shock protein 70 peptide complex derived from dendritic tumor fusion cells. Oncol. Rep. 2015, 33, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Chen, J.; Liu, Y.; Luo, W. Dendritic-tumor fusion cells derived heat shock protein70-Peptide complex has enhanced immunogenicity. PLoS ONE 2015, 10, e0126075. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Fucikova, J.; Garg, A.D.; Agostinis, P.; Špíšek, R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2014, 3, e968434. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.R.; Jammeh, M.L.; Wattenberg, M.M.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 2014, 5, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Combination of chemotherapy and immunotherapy for cancer: A paradigm revisited. Lancet Oncol. 2007, 8, 2–3. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Haanen, J.B.; Punt, C.J. Cancer immunotherapy—Revisited. Nat. Rev. Drug Discov. 2011, 10, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Shinkai, M.; Honda, H.; Yoshikawa, K.; Saga, S.; Wakabayashi, T.; Yoshida, J.; Kobayashi, T. Heat shock protein 70 expression induces antitumor immunity during intracellular hyperthermia using magnetite nanoparticles. Cancer Immunol. Immunother. 2003, 52, 80–88. [Google Scholar] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Kan, S.; Yoshida, K.; Yoshizaki, S.; Takakura, K.; Namiki, Y.; Tsukinaga, S.; Odahara, S.; Kajihara, M.; Okamoto, M.; et al. Immunogenic modulation of cholangiocarcinoma cells by chemoimmunotherapy. Anticancer Res. 2014, 34, 6353–6361. [Google Scholar] [PubMed]
- Takahara, A.; Koido, S.; Ito, M.; Nagasaki, E.; Sagawa, Y.; Iwamoto, T.; Komita, H.; Ochi, T.; Fujiwara, H.; Yasukawa, M.; et al. Gemcitabine enhances Wilms' tumor gene WT1 expression and sensitizes human pancreatic cancer cells with WT1-specific T-cell-mediated antitumor immune response. Cancer Immunol. Immunother. 2011, 60, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Koido, S.; Okamoto, M.; Hayashi, K.; Ito, M.; Kamata, Y.; Komita, H.; Ishidao, T.; Nagasaki, E.; Homma, S. Gemcitabine treatment enhances HER2 expression in low HER2-expressing breast cancer cells and enhances the antitumor effects of trastuzumab emtansine. Oncol. Rep. 2015, 34, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Koido, S.; Okamoto, M.; Hayashi, K.; Ito, M.; Kamata, Y.; Komita, H.; Nagasaki, E.; Homma, S. Up-regulation of HER2 by gemcitabine enhances the antitumor effect of combined gemcitabine and trastuzumab emtansine treatment on pancreatic ductal adenocarcinoma cells. BMC Cancer 2015, 15, 726. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Hara, E.; Mitsunaga, M.; Namiki, Y.; Takahara, A.; Nagasaki, E.; Komita, H.; Sagawa, Y.; Ohkusa, T.; et al. In vitro generation of cytotoxic and regulatory T cells by fusions of human dendritic cells and hepatocellular carcinoma cells. J. Transl. Med. 2008, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Hazama, S.; Maeda, K.; Inoue, Y.; Homma, S.; Koido, S.; Okamoto, M.; Oka, M. Suppressive effects of cyclophosphamide and gemcitabine on regulatory T-cell induction in vitro. Anticancer Res. 2012, 32, 5363–5369. [Google Scholar] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Vasir, B.; Tzachanis, D.; Levine, J.D.; Joyce, R.M.; Wellenstein, K.; Keefe, W.; Schickler, M.; et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J. Immunother. 2011, 34, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Reddy, V.; Dannull, J.; Ding, E.; Nair, S.K.; Tyler, D.S.; Pruitt, S.K.; Lee, W.T. Impact of anti-CD25 monoclonal antibody on dendritic cell-tumor fusion vaccine efficacy in a murine melanoma model. J. Transl. Med. 2013, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Avigan, D.; Rosenblatt, J.; Kufe, D. Dendritic/tumor fusion cells as cancer vaccines. Semin. Oncol. 2012, 39, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Fusion cells generated with dendritic cells and whole tumor cells. Fusions of dendritic cells (DCs) and whole tumor cells (DC-tumor FCs) display a characteristic phenotype comprised of major histocompatibility complex (MHC) class I molecules, MHC class II molecules, co-stimulatory molecules (CD80 and CD86), and multiple tumor-associated antigens. Multiple tumor-associated antigens from whole tumor cells are degraded by proteasome in DC-tumor FCs (solid line arrow), and antigenic peptides are loaded onto MHC class I molecules in the endoplasmic reticulum. The peptide-MHC class I complexes are then expressed on the DC-tumor FC surface (dash line arrow) and stimulate polyclonal antigen-specific CD8+ T cells. DC-tumor FCs can also synthesize MHC class II-restricted antigenic peptides in the endoplasmic reticulum. These are transported to the cytoplasm, where peptide-MHC class II complexes are generated (dash line arrow). Peptide-MHC class II complexes are also expressed on the DC-tumor FC surface and activate polyclonal antigen-specific CD4+ T cells.
Figure 1.
Fusion cells generated with dendritic cells and whole tumor cells. Fusions of dendritic cells (DCs) and whole tumor cells (DC-tumor FCs) display a characteristic phenotype comprised of major histocompatibility complex (MHC) class I molecules, MHC class II molecules, co-stimulatory molecules (CD80 and CD86), and multiple tumor-associated antigens. Multiple tumor-associated antigens from whole tumor cells are degraded by proteasome in DC-tumor FCs (solid line arrow), and antigenic peptides are loaded onto MHC class I molecules in the endoplasmic reticulum. The peptide-MHC class I complexes are then expressed on the DC-tumor FC surface (dash line arrow) and stimulate polyclonal antigen-specific CD8+ T cells. DC-tumor FCs can also synthesize MHC class II-restricted antigenic peptides in the endoplasmic reticulum. These are transported to the cytoplasm, where peptide-MHC class II complexes are generated (dash line arrow). Peptide-MHC class II complexes are also expressed on the DC-tumor FC surface and activate polyclonal antigen-specific CD4+ T cells.

Figure 2.
Fusion cells generated with autologous dendritic cells and allogeneic tumor cells. Fusions of autologous dendritic cells (Auto-DCs) and allogeneic tumor cells (Auto-DC/Allo-Tumor FC) display a characteristic phenotype comprised of major histocompatibility complex (MHC) class I molecules from autologous DCs and allogeneic tumor cells, MHC class II molecules from autologous DCs, co-stimulatory molecules (CD80 and CD86), and multiple tumor-associated antigens. Multiple tumor-associated antigens from allogeneic tumor cells are degraded by proteasome in Auto-DC/Allo-Tumor FC (solid line arrow), and antigenic peptides are loaded onto MHC class I molecules from autologous DC (black-colored dash line arrow) and allogeneic tumor cell (red-colored dash line arrow) in the endoplasmic reticulum. The peptide-MHC class I complexes are then expressed on the Auto-DC/Allo-Tumor FC surface and stimulate polyclonal antigen-specific CD8+ T cells and alloreactive CD8+ T cells. Auto-DC/Allo-Tumor FC can also synthesize MHC class II-restricted antigenic peptides in the endoplasmic reticulum. These are transported to the cytoplasm, where peptide-autologous MHC class II complexes are generated (black-colored dash line arrow). Peptide-autologous MHC class II complexes are also expressed on the Auto-DC/Allo-Tumor FC surface and activate polyclonal antigen-specific CD4+ T cells. Moreover, antigens derived from Auto-DC/Allo-Tumor FC are also be cross-presented by host DCs, resulting in the induction of polyclonal antigen-specific CD4+ and CD8+ T cells.
Figure 2.
Fusion cells generated with autologous dendritic cells and allogeneic tumor cells. Fusions of autologous dendritic cells (Auto-DCs) and allogeneic tumor cells (Auto-DC/Allo-Tumor FC) display a characteristic phenotype comprised of major histocompatibility complex (MHC) class I molecules from autologous DCs and allogeneic tumor cells, MHC class II molecules from autologous DCs, co-stimulatory molecules (CD80 and CD86), and multiple tumor-associated antigens. Multiple tumor-associated antigens from allogeneic tumor cells are degraded by proteasome in Auto-DC/Allo-Tumor FC (solid line arrow), and antigenic peptides are loaded onto MHC class I molecules from autologous DC (black-colored dash line arrow) and allogeneic tumor cell (red-colored dash line arrow) in the endoplasmic reticulum. The peptide-MHC class I complexes are then expressed on the Auto-DC/Allo-Tumor FC surface and stimulate polyclonal antigen-specific CD8+ T cells and alloreactive CD8+ T cells. Auto-DC/Allo-Tumor FC can also synthesize MHC class II-restricted antigenic peptides in the endoplasmic reticulum. These are transported to the cytoplasm, where peptide-autologous MHC class II complexes are generated (black-colored dash line arrow). Peptide-autologous MHC class II complexes are also expressed on the Auto-DC/Allo-Tumor FC surface and activate polyclonal antigen-specific CD4+ T cells. Moreover, antigens derived from Auto-DC/Allo-Tumor FC are also be cross-presented by host DCs, resulting in the induction of polyclonal antigen-specific CD4+ and CD8+ T cells.

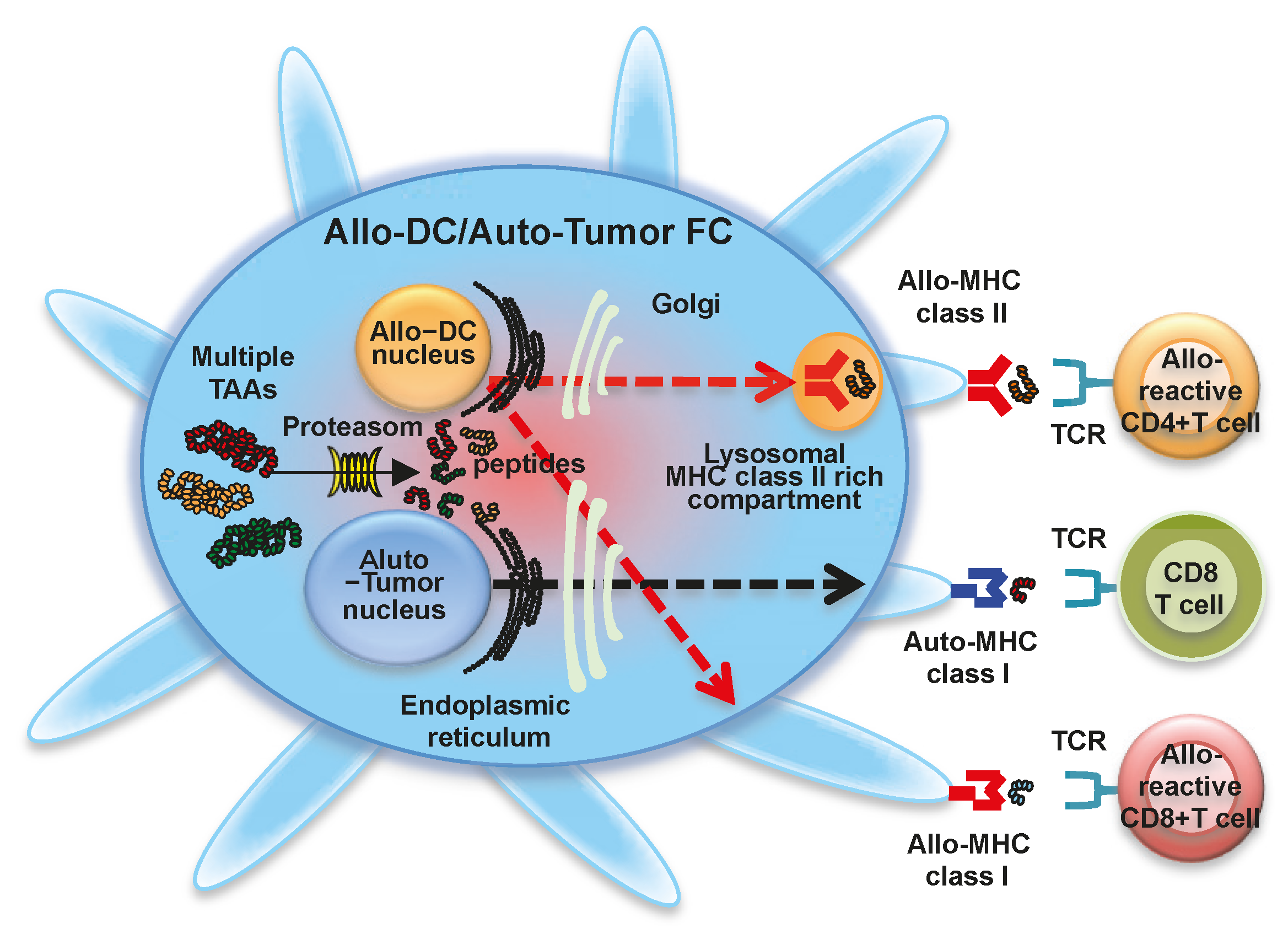
Figure 3.
Fusion cells generated with allogeneic dendritic cells and autologous tumor cells. Fusions of allogeneic dendritic cells (Allo-DCs) and autologous tumor cells (Allo-DC/Auto-Tumor FC) display a characteristic phenotype comprised of MHC class I molecules from allogeneic DCs and autologous tumor cells, MHC class II molecules from allogeneic DCs, co-stimulatory molecules (CD80 and CD86), and multiple tumor-associated antigens. Multiple tumor-associated antigens from autologous tumor cells are degraded by proteasome in Allo-DC/Auto-Tumor FC (solid line arrow), and antigenic peptides are loaded onto MHC class I molecules from allogeneic DC (red-colored dash line arrow) and autologous tumor cell (black-colored dash line arrow) in the endoplasmic reticulum. The peptide-MHC class I complexes are then expressed on the Allo-DC/Auto-Tumor FC surface and stimulate polyclonal antigen-specific CD8+ T cells and alloreactive CD8+ T cells. Allo-DC/Auto-Tumor FC can also synthesize MHC class II-restricted antigenic peptides in the endoplasmic reticulum. These are transported to the cytoplasm, where peptide-allogeneic MHC class II complexes are generated (red-colored dash line arrow). Peptide-allogeneic MHC class II complexes are also expressed on the Allo-DC/Auto-Tumor FC surface and activate alloreactive CD4+ T cells. Moreover, antigens derived from Allo-DC/Auto-Tumor FC are also be cross-presented by host DCs, resulting in the induction of polyclonal antigen-specific CD4+ and CD8+ T cells.
Figure 3.
Fusion cells generated with allogeneic dendritic cells and autologous tumor cells. Fusions of allogeneic dendritic cells (Allo-DCs) and autologous tumor cells (Allo-DC/Auto-Tumor FC) display a characteristic phenotype comprised of MHC class I molecules from allogeneic DCs and autologous tumor cells, MHC class II molecules from allogeneic DCs, co-stimulatory molecules (CD80 and CD86), and multiple tumor-associated antigens. Multiple tumor-associated antigens from autologous tumor cells are degraded by proteasome in Allo-DC/Auto-Tumor FC (solid line arrow), and antigenic peptides are loaded onto MHC class I molecules from allogeneic DC (red-colored dash line arrow) and autologous tumor cell (black-colored dash line arrow) in the endoplasmic reticulum. The peptide-MHC class I complexes are then expressed on the Allo-DC/Auto-Tumor FC surface and stimulate polyclonal antigen-specific CD8+ T cells and alloreactive CD8+ T cells. Allo-DC/Auto-Tumor FC can also synthesize MHC class II-restricted antigenic peptides in the endoplasmic reticulum. These are transported to the cytoplasm, where peptide-allogeneic MHC class II complexes are generated (red-colored dash line arrow). Peptide-allogeneic MHC class II complexes are also expressed on the Allo-DC/Auto-Tumor FC surface and activate alloreactive CD4+ T cells. Moreover, antigens derived from Allo-DC/Auto-Tumor FC are also be cross-presented by host DCs, resulting in the induction of polyclonal antigen-specific CD4+ and CD8+ T cells.

Figure 4.
Fusion cells generated with allogeneic dendritic cells and allogeneic tumor cells. Fusions of allogeneic dendritic cells (Allo-DCs) and allogeneic tumor cells (Allo-DC/Allo-Tumor FC) display a characteristic phenotype comprised of MHC class I molecules from allogeneic DCs and allogeneic tumor cells, MHC class II molecules from allogeneic DCs, co-stimulatory molecules (CD80 and CD86), and multiple tumor-associated antigens. Multiple tumor-associated antigens from allogeneic tumor cells are degraded by proteasome in Allo-DC/Allo-Tumor FC (solid line arrow), and antigenic peptides are loaded onto MHC class I molecules from allogeneic DC (red-colored dash line arrow) and allogeneic tumor cell (red-colored dash line arrow) in the endoplasmic reticulum. The peptide-MHC class I complexes are then expressed on the Allo-DC/Allo-Tumor FC surface and stimulate alloreactive CD8+ T cells. Allo-DC/Allo-Tumor FC can also synthesize MHC class II-restricted antigenic peptides in the endoplasmic reticulum. These are transported to the cytoplasm, where peptide-allogeneic MHC class II complexes are generated (red-colored dash line arrow). Peptide-allogeneic MHC class II complexes are also expressed on the Allo-DC/Allo-Tumor FC surface and activate alloreactive CD4+ T cells. Moreover, antigens derived from Allo-DC/Allo-Tumor FC are also be cross-presented by host DCs, resulting in the induction of polyclonal antigen-specific CD4+ and CD8+ T cells.
Figure 4.
Fusion cells generated with allogeneic dendritic cells and allogeneic tumor cells. Fusions of allogeneic dendritic cells (Allo-DCs) and allogeneic tumor cells (Allo-DC/Allo-Tumor FC) display a characteristic phenotype comprised of MHC class I molecules from allogeneic DCs and allogeneic tumor cells, MHC class II molecules from allogeneic DCs, co-stimulatory molecules (CD80 and CD86), and multiple tumor-associated antigens. Multiple tumor-associated antigens from allogeneic tumor cells are degraded by proteasome in Allo-DC/Allo-Tumor FC (solid line arrow), and antigenic peptides are loaded onto MHC class I molecules from allogeneic DC (red-colored dash line arrow) and allogeneic tumor cell (red-colored dash line arrow) in the endoplasmic reticulum. The peptide-MHC class I complexes are then expressed on the Allo-DC/Allo-Tumor FC surface and stimulate alloreactive CD8+ T cells. Allo-DC/Allo-Tumor FC can also synthesize MHC class II-restricted antigenic peptides in the endoplasmic reticulum. These are transported to the cytoplasm, where peptide-allogeneic MHC class II complexes are generated (red-colored dash line arrow). Peptide-allogeneic MHC class II complexes are also expressed on the Allo-DC/Allo-Tumor FC surface and activate alloreactive CD4+ T cells. Moreover, antigens derived from Allo-DC/Allo-Tumor FC are also be cross-presented by host DCs, resulting in the induction of polyclonal antigen-specific CD4+ and CD8+ T cells.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Advantages | DC-tumor FCs present whole tumor-derived antigenic peptides, which avoids the need to identify antigenic peptides for individual patients. |
| A broad array of known and unidentified tumor-associated antigens are simultaneously presented on the surface of DC-tumor FCs. | |
| Endogenously-synthesized tumor-associated antigens in DC-tumor FCs are better access to MHC class I and II molecules. | |
| Increased the frequency of polyclonal antigen-specific CD4+ and CD8+ T cells can be induced by DC-tumor FCs. | |
| DC-tumor FCs can induce long-term efficient antitumor immunity. | |
| Numerous tumor-associated antigens are presented in the context of co-stimulatory molecules in DC-tumor FCs. | |
| DC-tumor FCs prevent tolerance induction. | |
| Autologous DC-autologous tumor FCs do not have to take up exogenous TAAs in order to activate CD4+ and CD8+ T cells. | |
| Modifications of DCs and tumor cells are independently possible while their characters present after the fusion. | |
| Allogeneic DC and allogeneic tumor cells can be used instead of autologous cells in generation of DC-tumor FCs. | |
| DC-tumor FC-based cancer vaccines can be combined with standard therapies. | |
| Disadvantages | The limited availability of viable autologous tumor cells as a fusion partner. |
| Induction of antigen-specific CD4+ and CD8+ T cell responses by allogeneic DC-tumor FCs are at least partly associated with sharing of MHC class I. | |
| Fusion efficiency depends on cell conditions due to the sensitivity of cells to PEG treatment. |
DC-tumor FCs: fusions of dendritic cells and whole tumor cells; MHC: Major histocompatibility complex; PEG: Polyethylene glycol.
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Koido, S. Dendritic-Tumor Fusion Cell-Based Cancer Vaccines. Int. J. Mol. Sci. 2016, 17, 828. https://doi.org/10.3390/ijms17060828
AMA Style
Koido S. Dendritic-Tumor Fusion Cell-Based Cancer Vaccines. International Journal of Molecular Sciences. 2016; 17(6):828. https://doi.org/10.3390/ijms17060828
Chicago/Turabian StyleKoido, Shigeo. 2016. "Dendritic-Tumor Fusion Cell-Based Cancer Vaccines" International Journal of Molecular Sciences 17, no. 6: 828. https://doi.org/10.3390/ijms17060828
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.