2.1. Simulation
To check the performance of the proposed model, we conducted a simulation study. We generated SNP data by bootstrapping samples focusing on gene
NCOA5 (see the real data analysis section for details about this gene). There are 1126 individuals and 15 SNPs in gene
NCOA5. By bootstrapping, we assumed the original sample is the population, then randomly sampled individuals with replacement with size
each time. During bootstrap, all the SNP data and the mother’s glucose level (
U) in each individual were drawn together as a vector. By doing so, we can maintain the LD structures among SNPs as well as the correlations between SNPs and
U. The response
Y was then generated from the following model:
where
and
,
, are the estimators of
and
based on the real data for gene
NCOA5,
is the
kth sparse PC in the bootstrapped samples with size
, and
ε is the error term following a normal distribution with mean 0 and variance
, where
c is a constant controlling the size of the variance, and
is the estimated variance in real data based on gene
NCOA5.
τ is a constant to control the effect size of the model. When
, we can assess the empirical false positive rate. When
, we can assess the testing power and we expect the power increases as
τ increases. We set the bootstrap sample size as
, and the constant
to check the finite sample performance of the proposed method. Specifically, we were interested in evaluating the false positive control and the power of detecting association under different sample sizes and error variances.
As a comparison, we also analyzed the data with the VC-PCR model (
4), and a simple linear regression model with a linear G×E interaction form,
i.e.,
where
are the 15 SNPs in gene
NCOA5, and
ε is an error following a normal distribution with mean 0 and finite variance. We conducted the overall SNP effect test by testing:
and
, and the SNP
interaction effect test by testing
. Let
,
,
,
, and
. Denote LRT for testing
by
, and LRT for testing
by
, where
is the log-likelihood under
,
is the log-likelihood under
, and
is the log-likelihood under the full model. The LTRs
and
asymptotically follow a
distribution with 30 and 15 degrees of freedom, respectively.
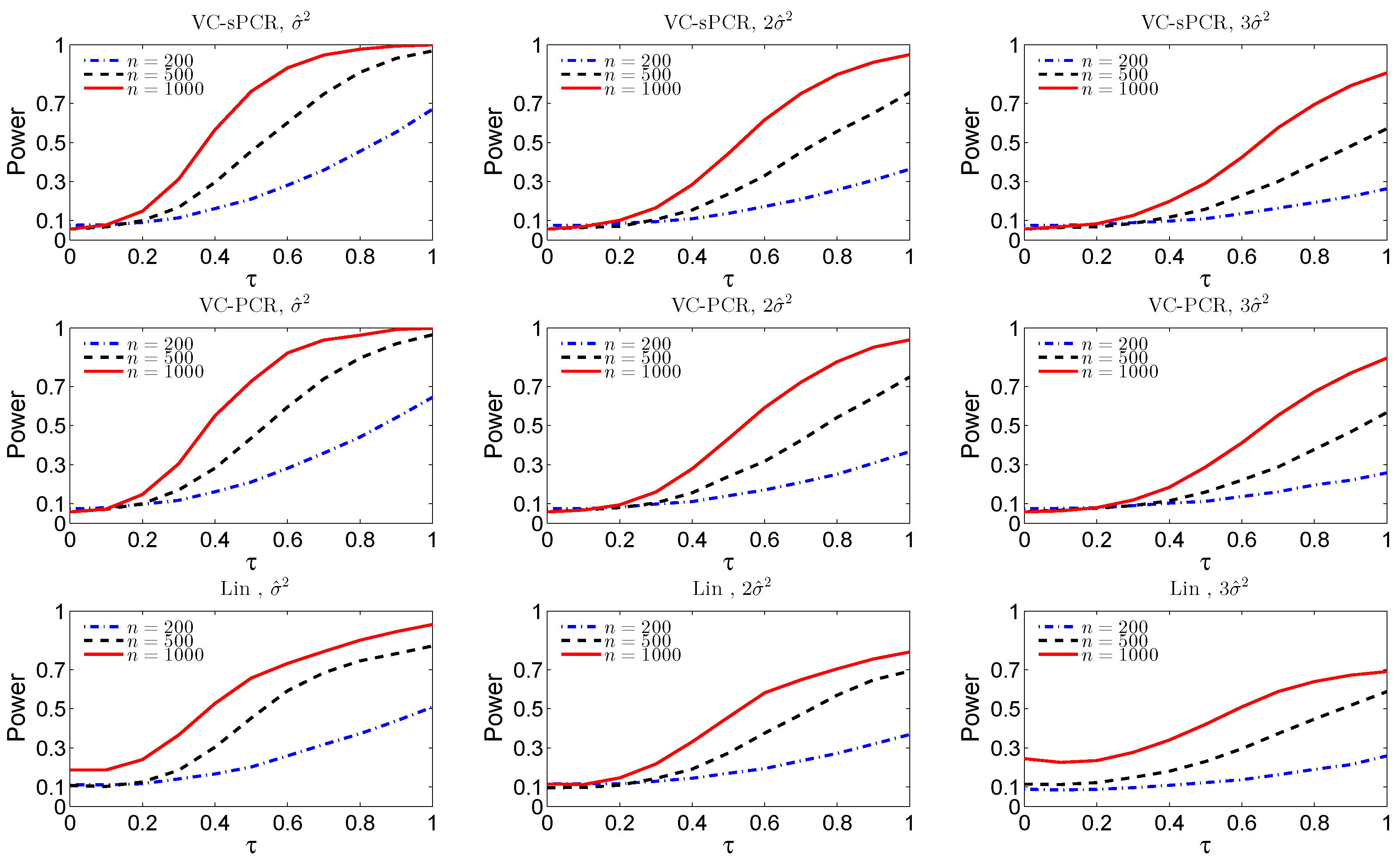
Figure 1 displays the empirical size (
) and power functions (
) under different sample sizes and error variances for the overall genetic effect test. The top three plots are for the overall genetic effect test fitted with the VC-sPCR model (
5), the middle three plots are for the overall genetic effect test fitted with the VC-PCR model (
4), and the bottom three plots are for the overall genetic effect test fitted with the linear G×E interaction model (
1). As we expected, the power and size improve as the sample size increases and the error variance decreases for all the three models. For both VC-sPCR and VC-PCR models, the size and power show very similar patterns. However, since sPCR analysis assumes sparsity of the PC loadings, hence has better interpretation. As a comparison, the linear regression model has the worst performance. First of all, the size is inflated and it gets worse when sample size increases, indicating completely failing of the linear model. This is expected since the simulated interaction function is nonlinear. Moreover, the power under the linear model is also worse than the other two models. We also simulated data assuming a linear G×E interaction effect. The results show that the performance of the VC-sPCR and VC-PCR models are very similar, but their performance is slightly worse than the results by fitting a linear G×E interaction model (data not shown due to space limit). A similar phenomenon was also observed in the original nonlinear G×E interaction model [
7]
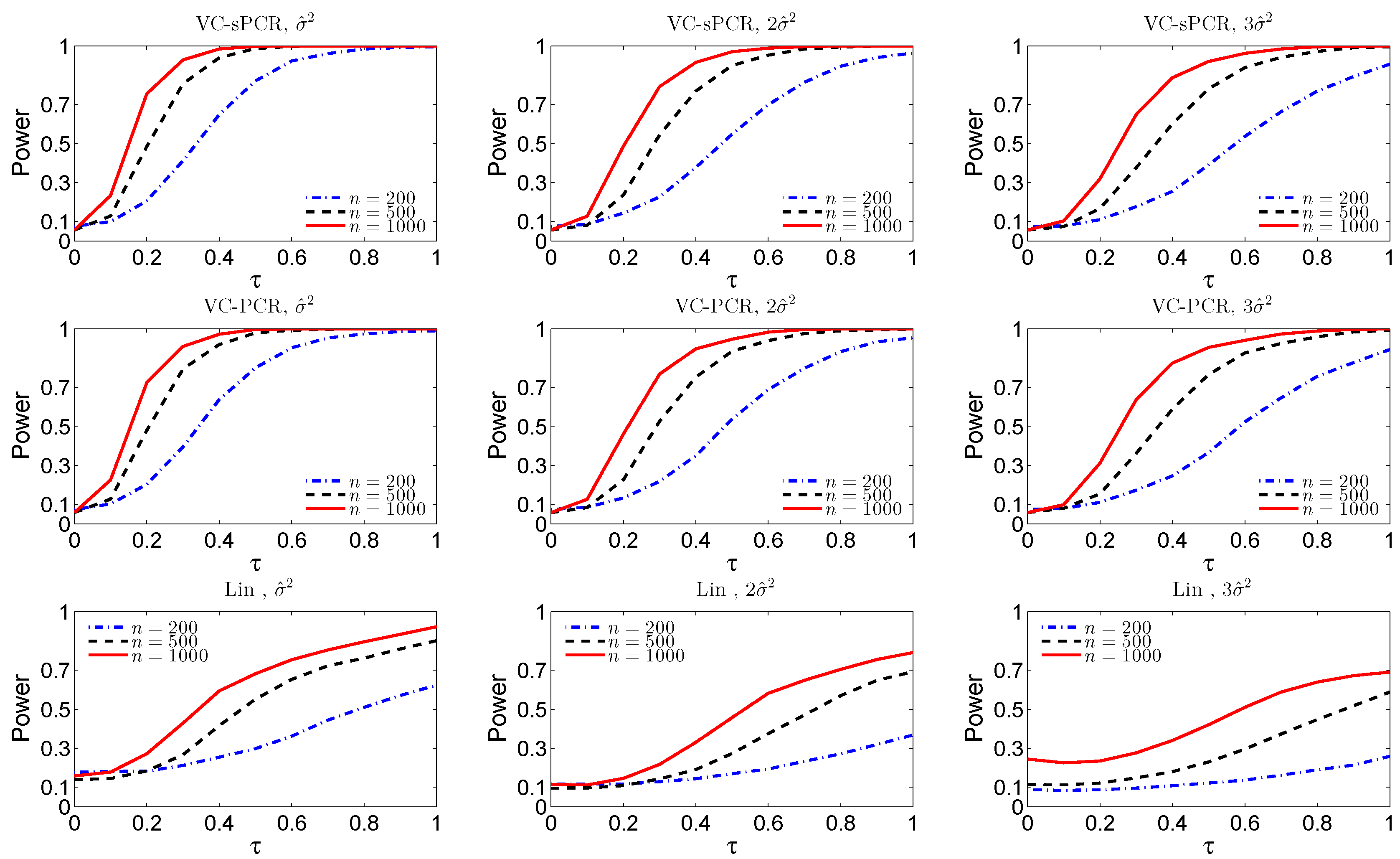
For the interaction test,
Figure 2 displays the empirical size and power functions under model VC-sPCR (
5) the first row, under model VC-PCR (
4) in the middle row, and under the linear G×E interaction model (
1) in the bottom row. Again, we observed very similar patterns as for the overall genetic effect test shown in
Figure 1.
2.2. Real Data Analysis
We applied the proposed model to a data set from the Gene Environment Association Studies initiative (GENEVA) funded by the trans-NIH (National Institute of Health) Genes, Environment, and Health Initiative (GEI), to identify important genes associated with birth weight. Fetal growth is not only determined by fetal genes but also controlled by complex interactions between fetal genes and the maternal uterine environment. In this example, we focused on the Thai population with 1126 subjects genotyped with the Omni1-Quad_v1-0_B platform (Illumina, San Diego, CA, USA) after removing potential outliers. We chose mother’s one hour OGTT (oral glucose tolerance test) glucose level (denoted as U) as the environmental variable in our analysis. Hypothetically, glucose from mothers can have big influence on fetal growth and such an effect can be partially captured by modeling the interaction mechanism between fetal genes and the glucose level coming from the mother.
There are total 590,913 SNPs after filtering out SNPs with minor allele frequency (MAF) <0.05, missing rate <0.05 and those deviating from Hardy–Weinberg equilibrium (
p-value
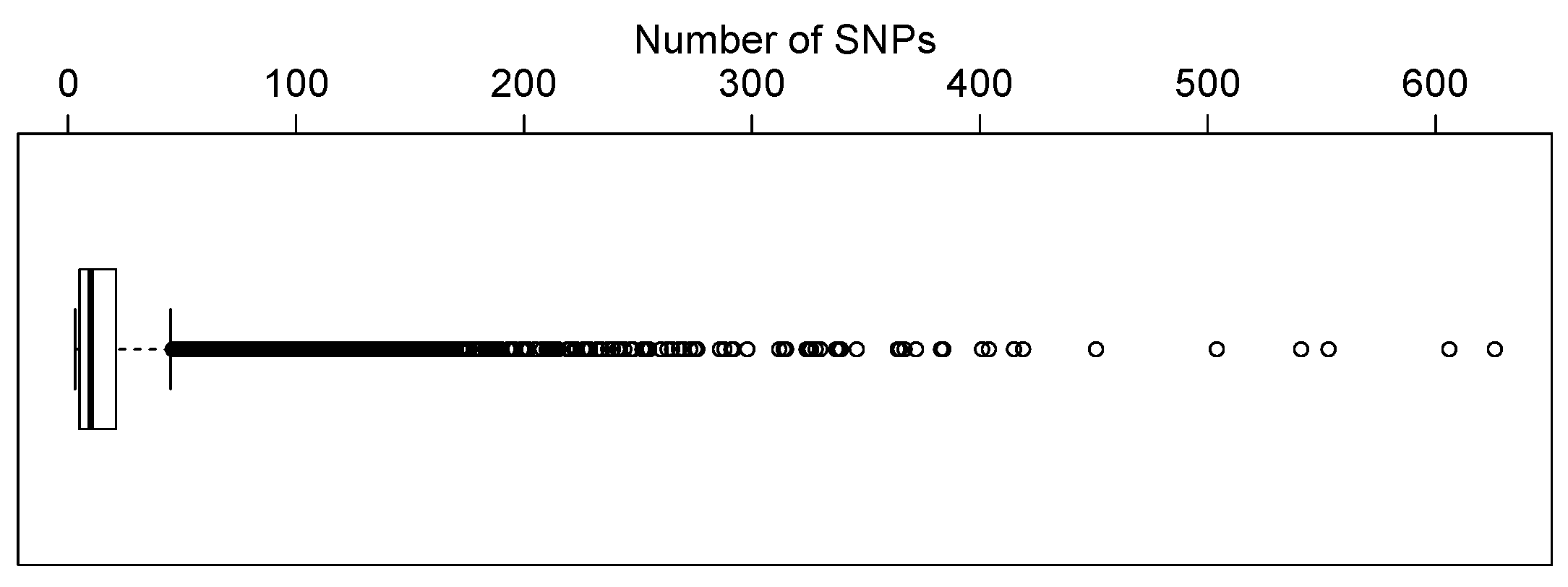
). These SNPs were then mapped to genes based on human genome builder 37 (GRCh37). We only focused on genes containing three or more SNPs in our analysis. This resulted in 12,005 genes. There are three genes containing relatively large number of SNPs (1355, 924, and 804 SNPs).
Figure 3 shows the distribution of the number of SNPs in genes by excluding these three genes. The number of SNPs in most genes is less than 20.
We centered the response first by subtracting the sample mean, then fitted the proposed varying-coefficient sparse PCR (VC-sPCR) model described in an earlier section to each gene, and conducted the aforementioned hypothesis testing. The number of PCs was chosen in such a way that >80% of gene variance can be explained by these PCs. As a comparison, we also fitted a regular varying-coefficient PCR (VC-PCR) model without assuming sparsity of the PC loadings. We first tested
to assess the overall genetic effect. The corresponding
p-values are denoted by
for VC-PCR model and
for VC-sPCR model (see
Table 1).
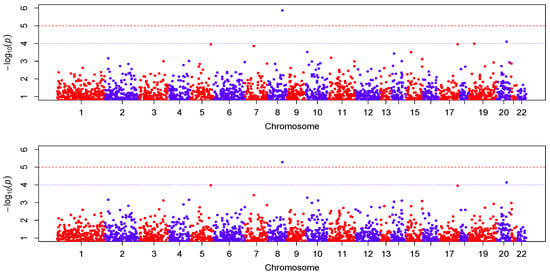
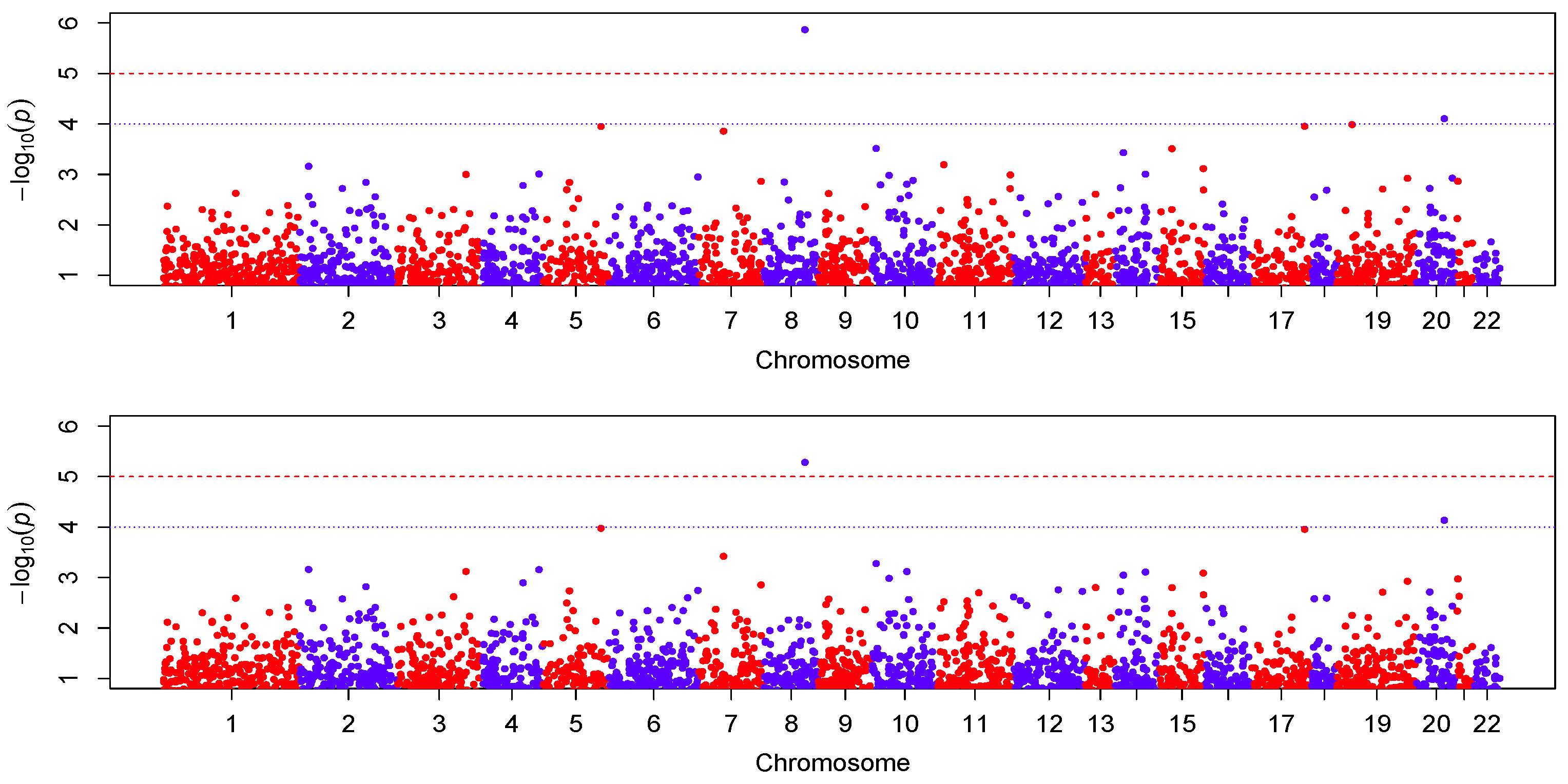
Figure 4 shows the Manhattan plot of the
p-values for the two models. The top panel is for the results analyzed with the VC-PCR model, and the bottom panel is for the results analyzed with the VC-sPCR model. The vertical axis is the −log10(
p-value) and horizontal axis shows the genes in 22 autosome chromosomes. The two models give quite consistent signals across all the genes. If we applied a Bonferroni threshold (−log10(0.05/12005) = 5.38) at a 0.05 genome-wide significance level, only one gene (
ANGPT1) on chromosome 8 shows significance. If we lowered the threshold to 1 ×
, then one gene (
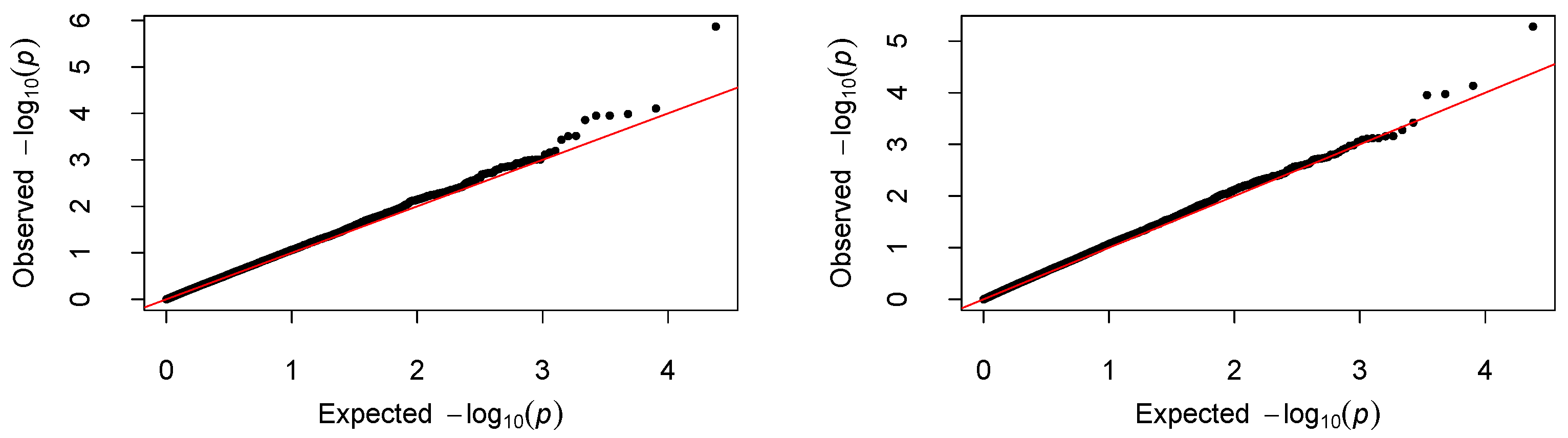
NCOA5) on chromosome 20 shows suggestive significance. The QQ-plots of the
p-values for the two models are given in
Figure 5. As we can see that no obvious departure from the diagonal line is observed, indicating no inflation of the
p-values.
Table 1 lists the two genes along with the gene name (Gene), chromosome (Chr), the number of PCs (
), the number of SNPs (
) within each gene, and the
p-values of different tests. The
p-values for testing
are denoted by
for the VC-PCR model and
for the VC-sPCR model, and the
p-values for testing
are denoted by
and
, respectively. The test results indicate that both the main and G×E interaction effects are significant for the two genes. For gene
NCOA5, the G×E interaction effect is stronger (
p-value = 1.5 ×
) than the main effect (
p-value = 3.29 ×
).
For those sparse PCs (sPCs) in each gene, we further tested the significance of each sPC. The results show that three out of seven sPCs are significant for gene
ANGPT1, and three out of four sPCs are significant for gene
NCOA5. The sparse PCs along with the
p-values, and the loadings are given in the
Supplementary File. In the file, we also listed those 67 SNPs in gene
ANGPT1 and 15 SNPs in gene
NCOA5 along with the sparse loadings for each SNP.
As we illustrated earlier, the proposed sparse PCs can ease the interpretation given the sparse loadings of the PCs. We conducted a single SNP test by fitting the following linear model,
where
is the SNP variable assuming an additive coding. We tested the total SNP effect by testing:
and the SNP×E interaction effect by testing
. The corresponding
p-values using a likelihood ratio test are denoted as
and
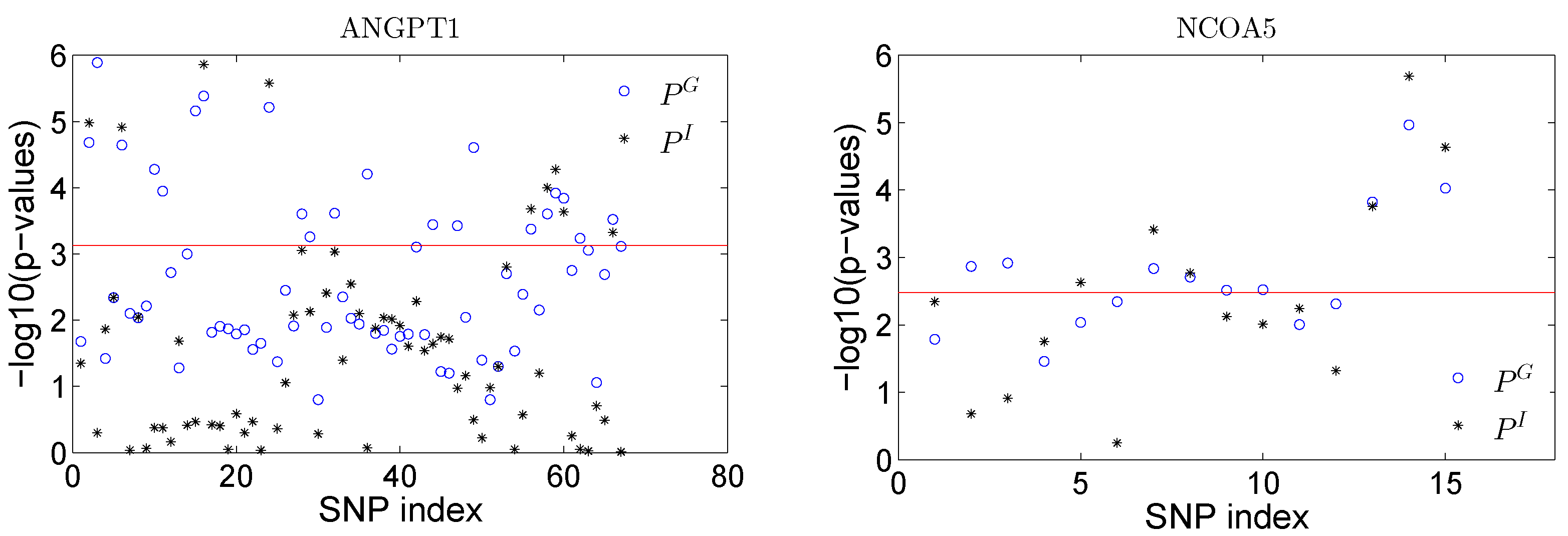
and are plotted in
Figure 6 for all SNPs in both genes. We can obtain some insights about the significant sPCs from the results. Take gene
NCOA5 as an example: the testing of a single sPC shows that PC1, PC2 and PC4 are significant at the 0.05 significance level. When checking the loadings for PC4, only the last three SNPs have large loadings, and the other SNPs have zero loadings on this PC. Thus, these three SNPs can be represented by PC4. A single SNP test shows that these three SNPs have the strongest effect among all the SNPs in terms of both overall and interaction effects (see
Figure 6). By the sparse representation of the PCs, we have nice interpretation about the significance of these sPCs. Note that the single SNP analysis conducted here is trying to illustrate the idea of the proposed sPCR analysis and to demonstrate whether non-zero loadings make any practical sense in real analysis.
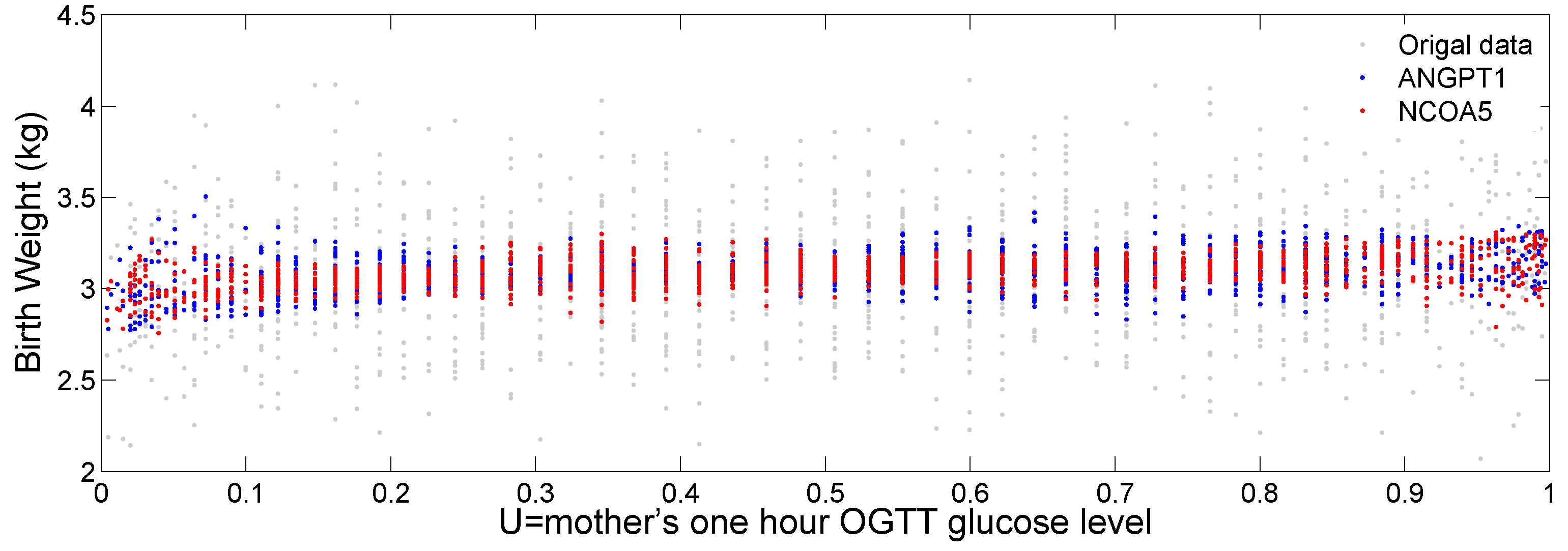
Figure 7 plots the original birth weight (grey dots), the fitted birth weight for gene
ANGPT1 (blue dots) and
NCOA5 (red dots) against the transformed glucose level (
U). We can see a slightly increasing trend of fitted birth weight for the two genes as the mother’s glucose level increases. We can also see a varying pattern of the fitted values against
U, indicating potential nonlinear interaction of the two genes with the mother’s glucose level to affect baby’s birth weight.
Gene
ANGPT1 encodes a secreted glycoprotein that belongs to the angiopoietin family and plays an important role in vascular development and angiogenesis. SNP rs2507800 in this gene has been shown to be associated with low birth weight and small-for-gestational-age infants [
12]. In our analysis, this SNP did not show significant association with birth weight. This might be due to the genetic heterogeneity of birth weight for the analyzed Thai population. However, our gene-based interaction model did find evidence of association at the gene level with birth weight. Gene
NCOA5 has been shown to be associated with diabetes [
13]. Many studies have shown that children born with low birth weight are associated with increased risk of developing type 2 diabetes (T2D) in adulthood [
14]. Several GWAS studies have identified genetic factors associated with T2D and birth weight (e.g., [
15]). As our analysis is focused on the Thai population, different from the previous GWAS report, it is possible that this gene shows significance only in the Thai population. Further studies are need to confirm this result. In addition, since we only analyzed genes containing more than two SNPs, we did not have a comprehensive coverage of all the SNP variants in our analysis. Thus, we could miss potential signals reported in other work simply because we did not analyze those variants. Our gene-based interaction analysis indicates the potential importance of these two genes on birth weight. Follow up studies will be conducted to verify the role of these two genes in other populations.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}