
Theoretical Studies on Structures, Properties and Dominant Debromination Pathways for Selected Polybrominated Diphenyl Ethers


Abstract
:
1. Introduction
2. Results and Discussion
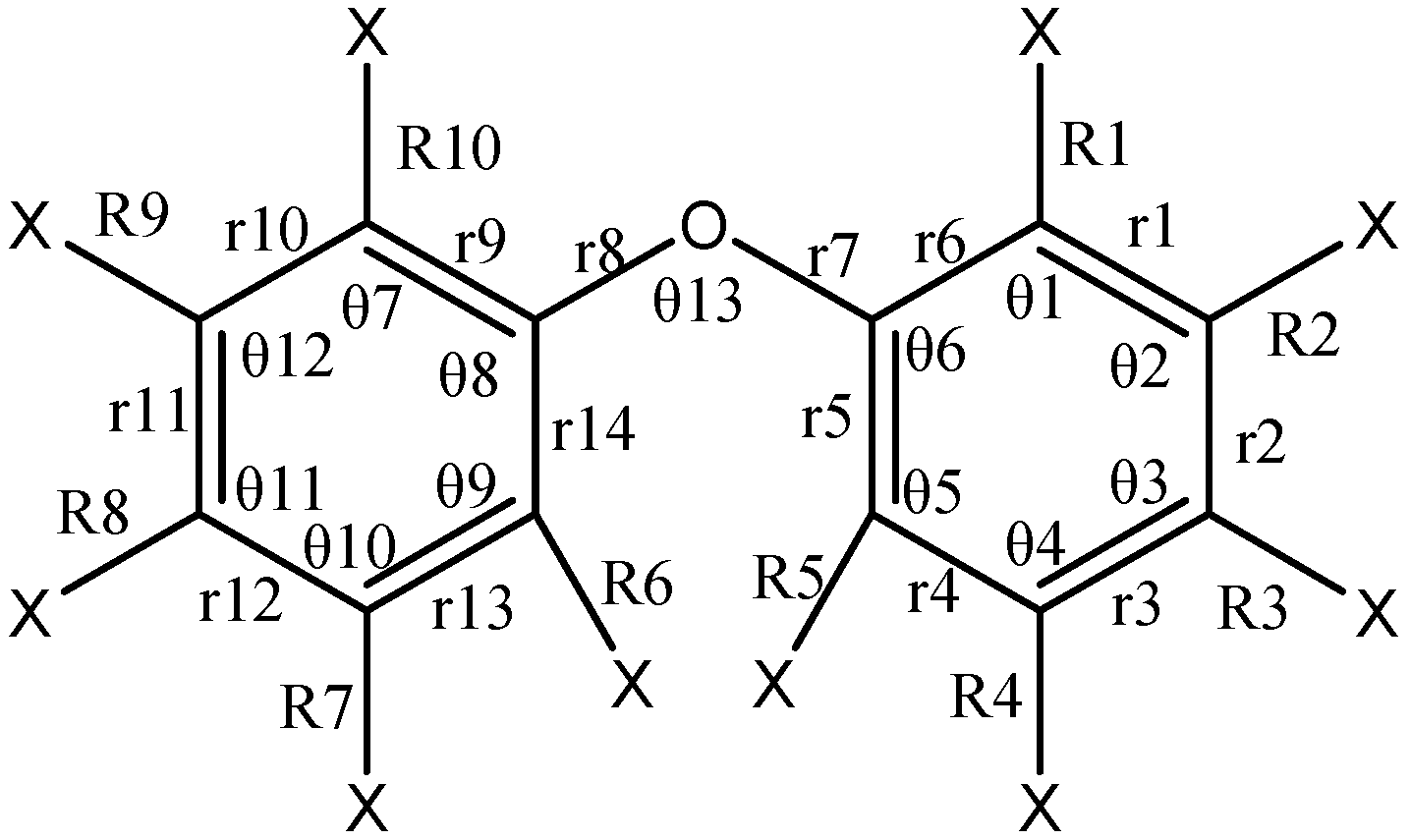
2.1. Molecular Geometry of Selected PBDEs
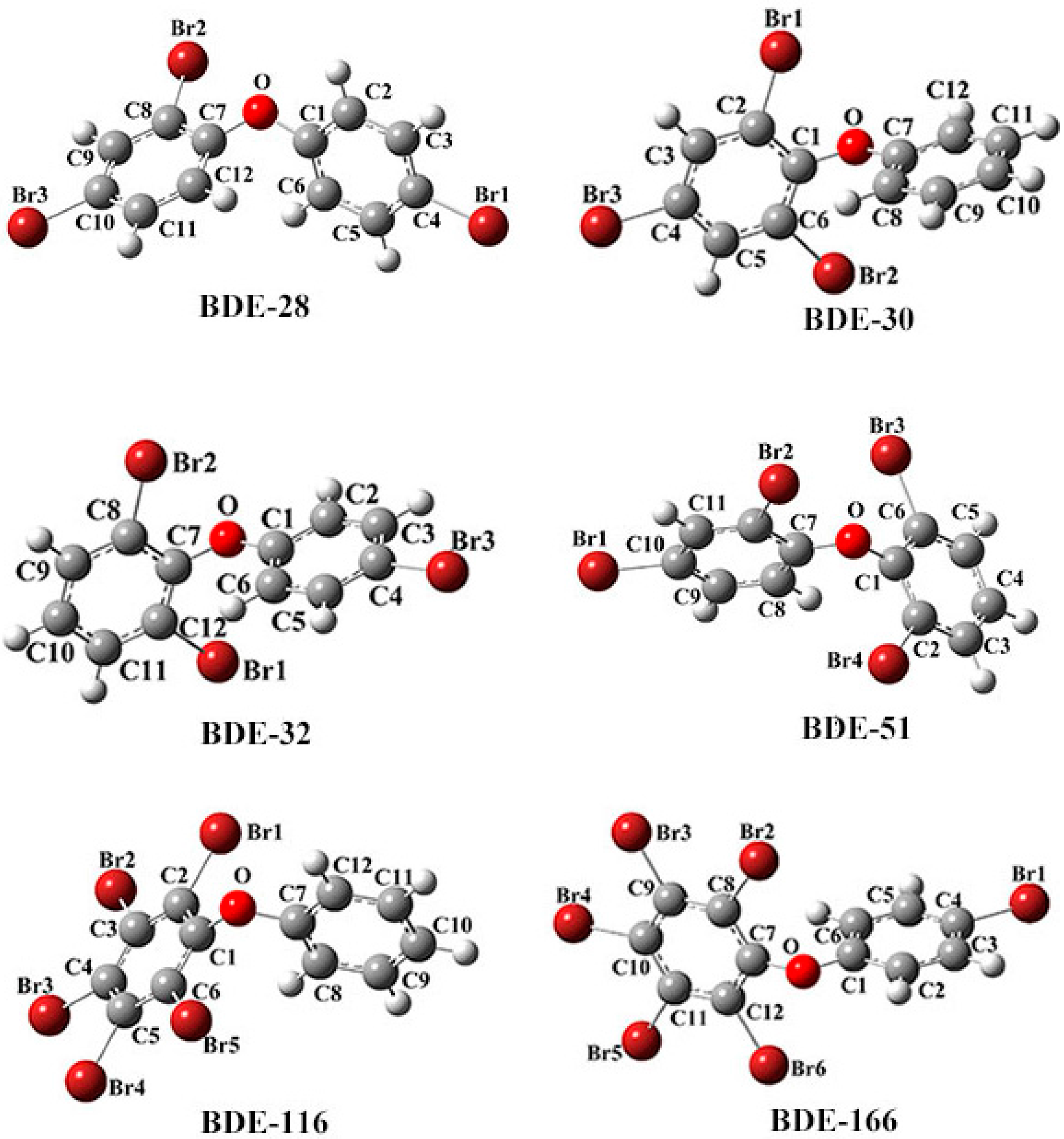
2.1.1. Comparison of Calculated Structural Parameters with Experimental Values for PBDEs
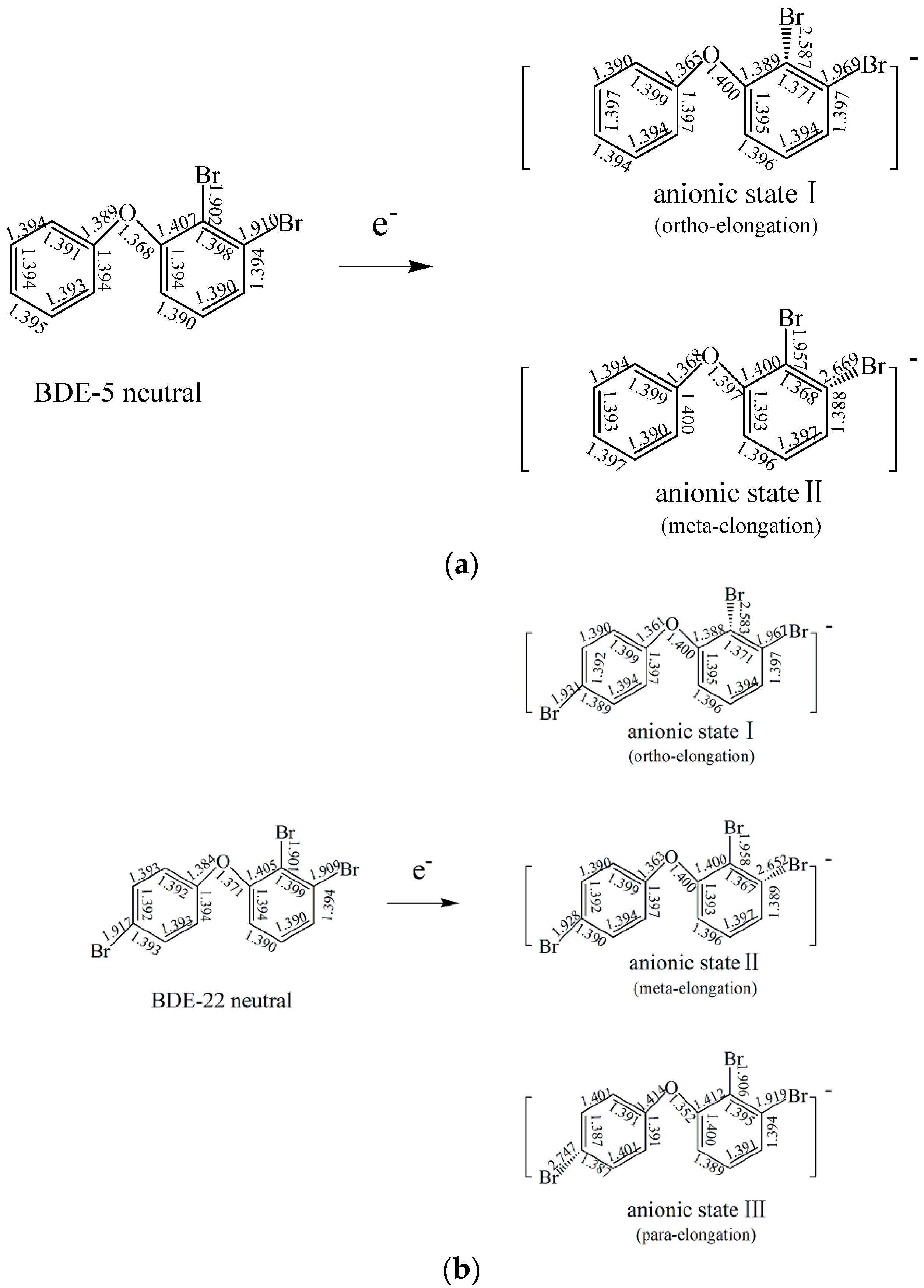
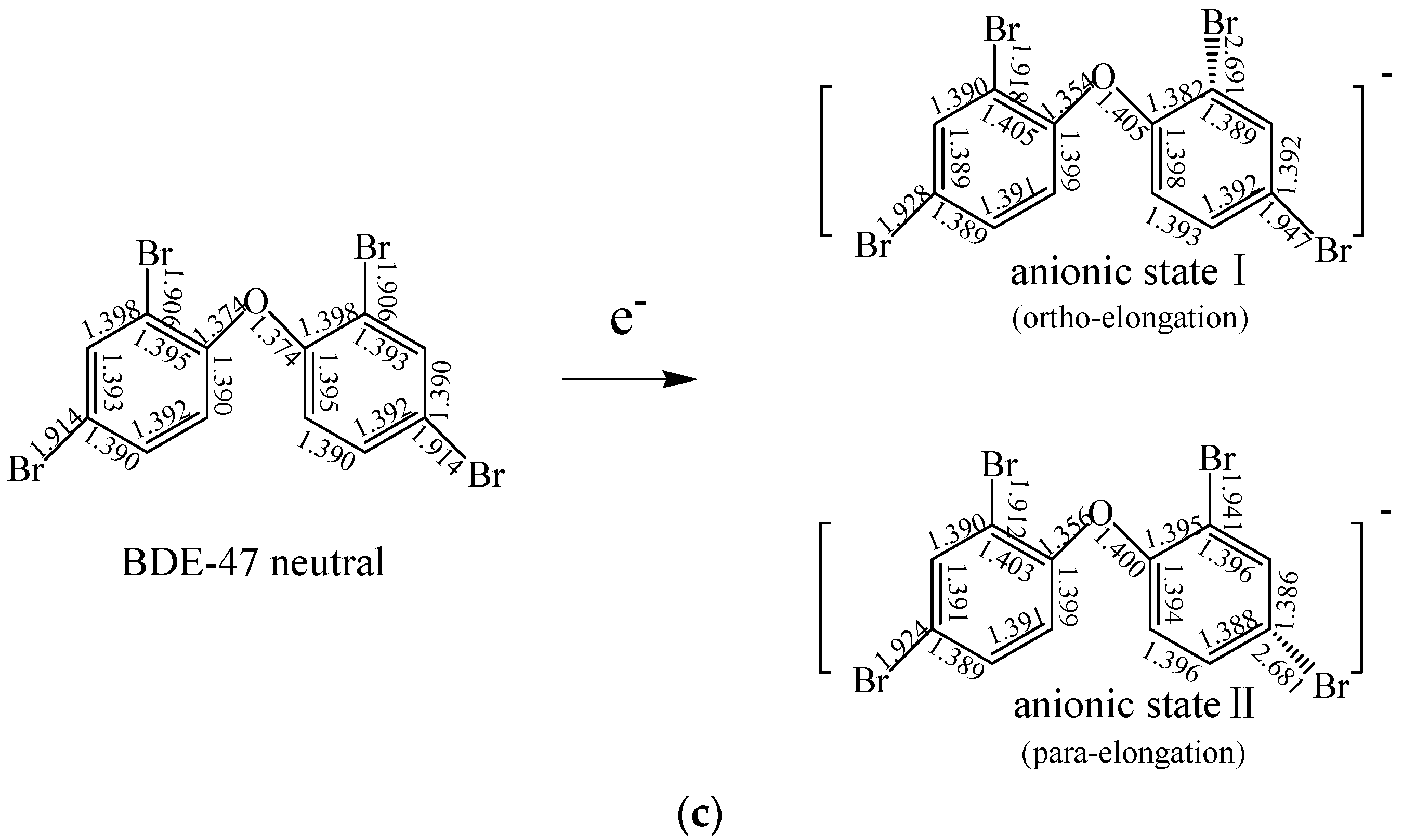
2.1.2. The Comparison of Structural Parameters between Neutral and Anionic Species of the Selected PBDEs
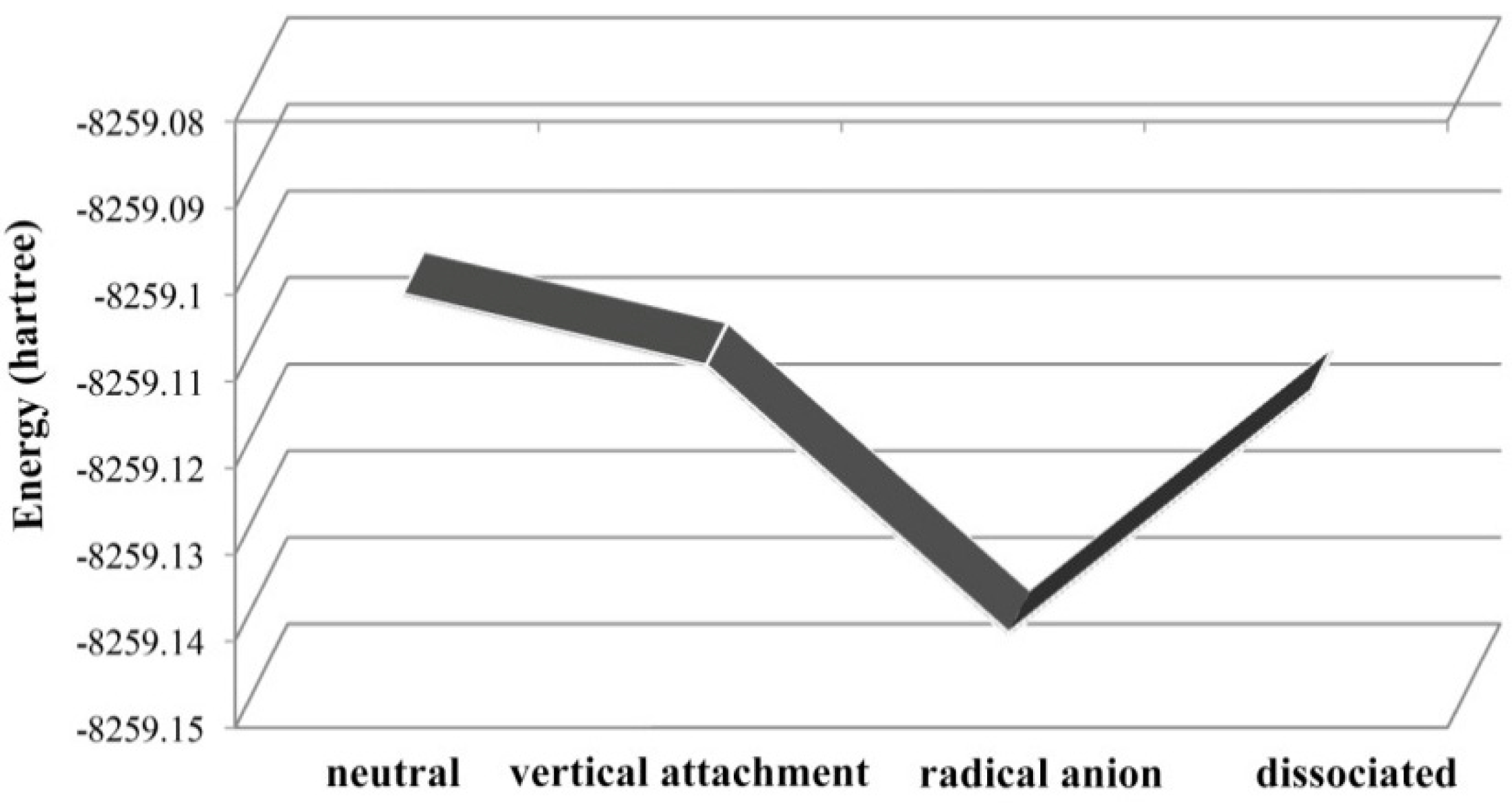
2.2. Electron Affinities (EA) of the Selected PBDEs
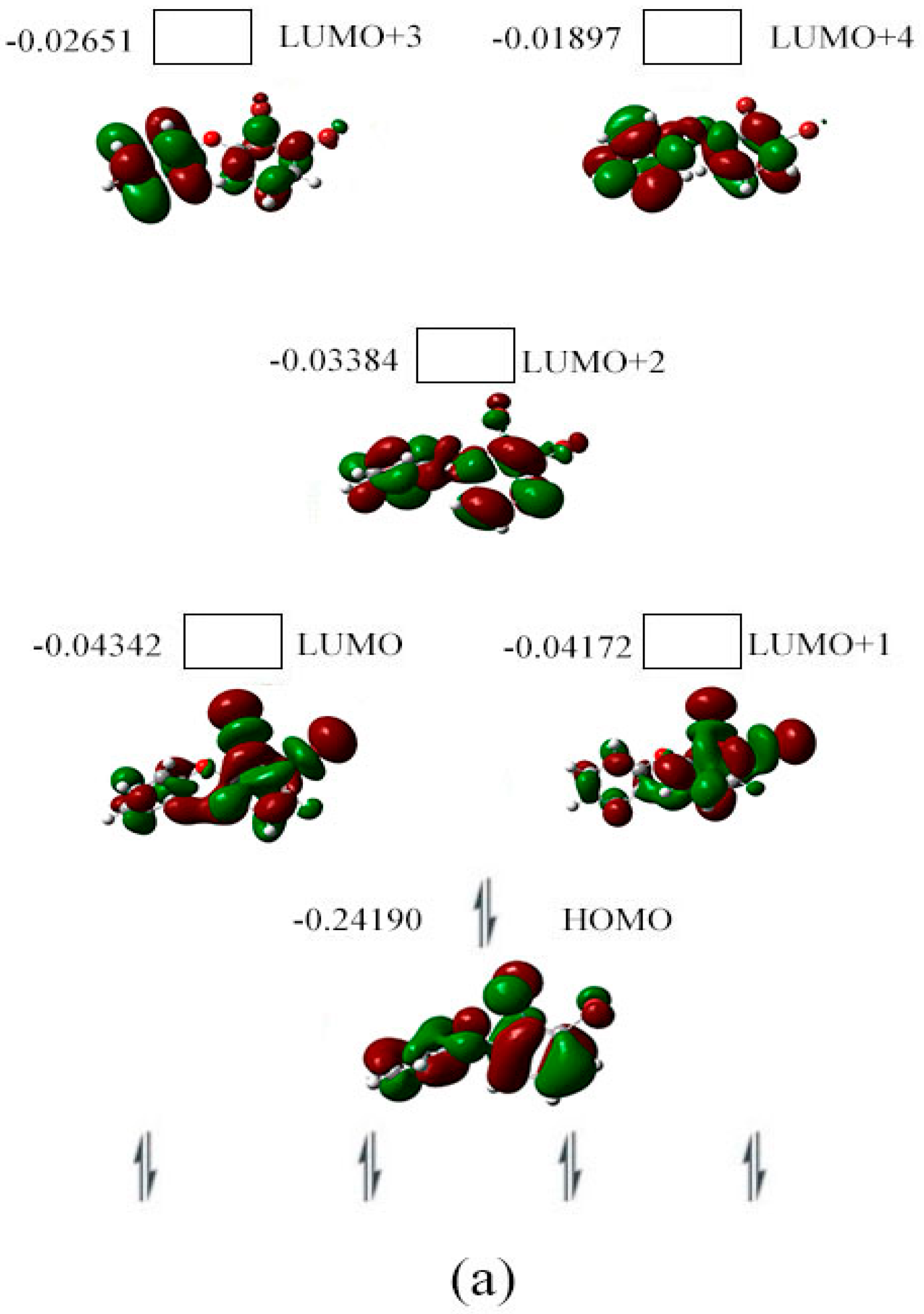
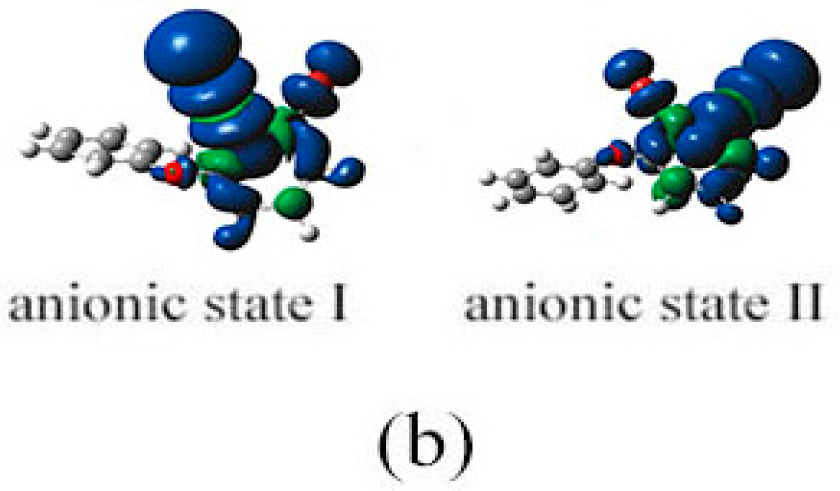
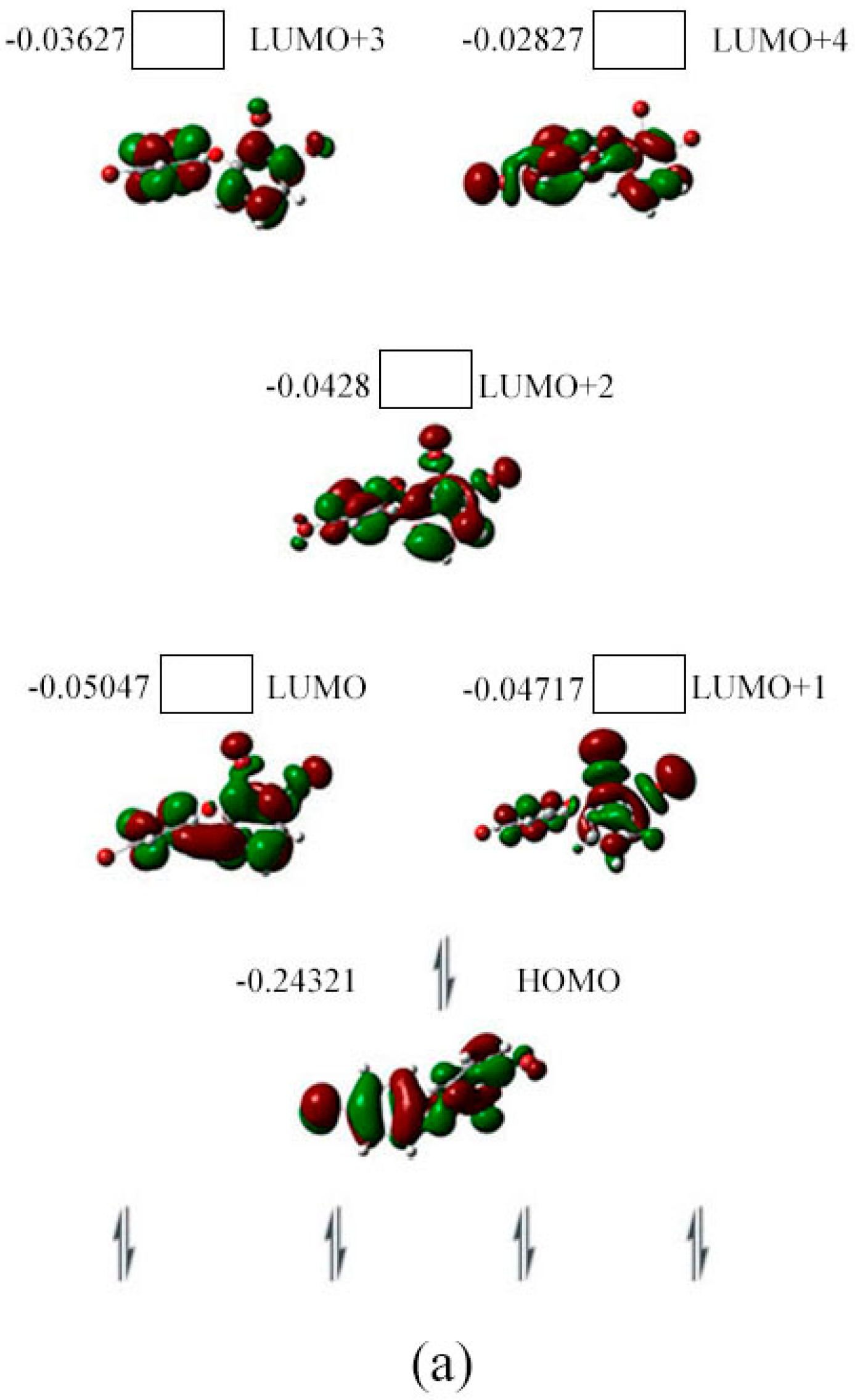
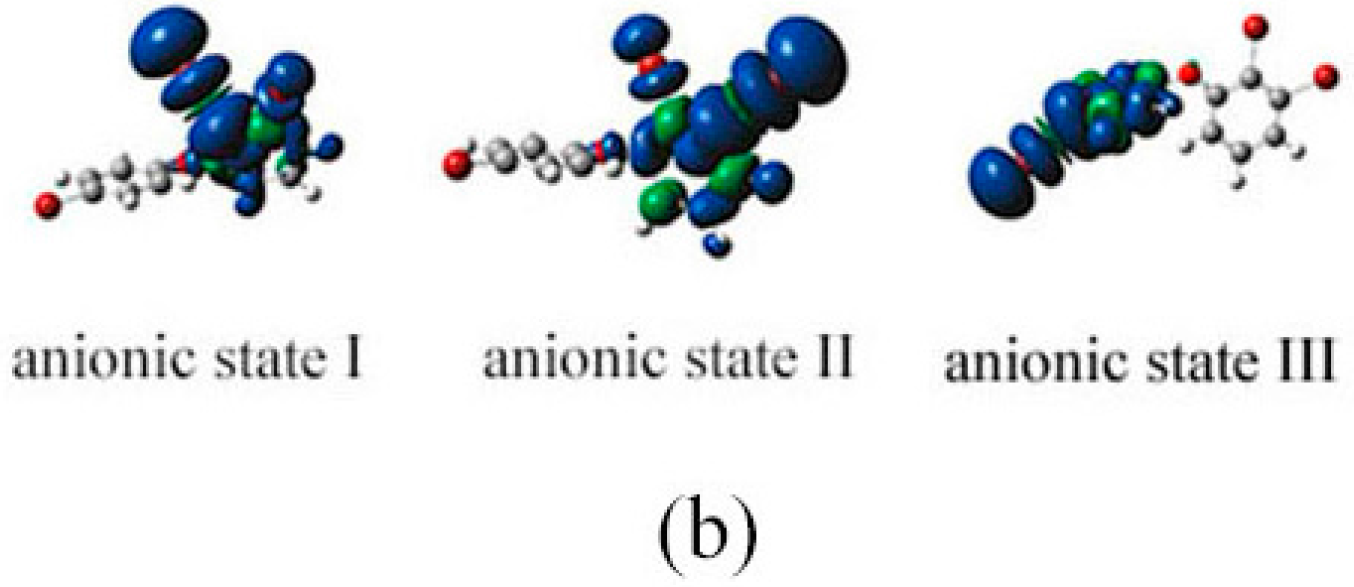
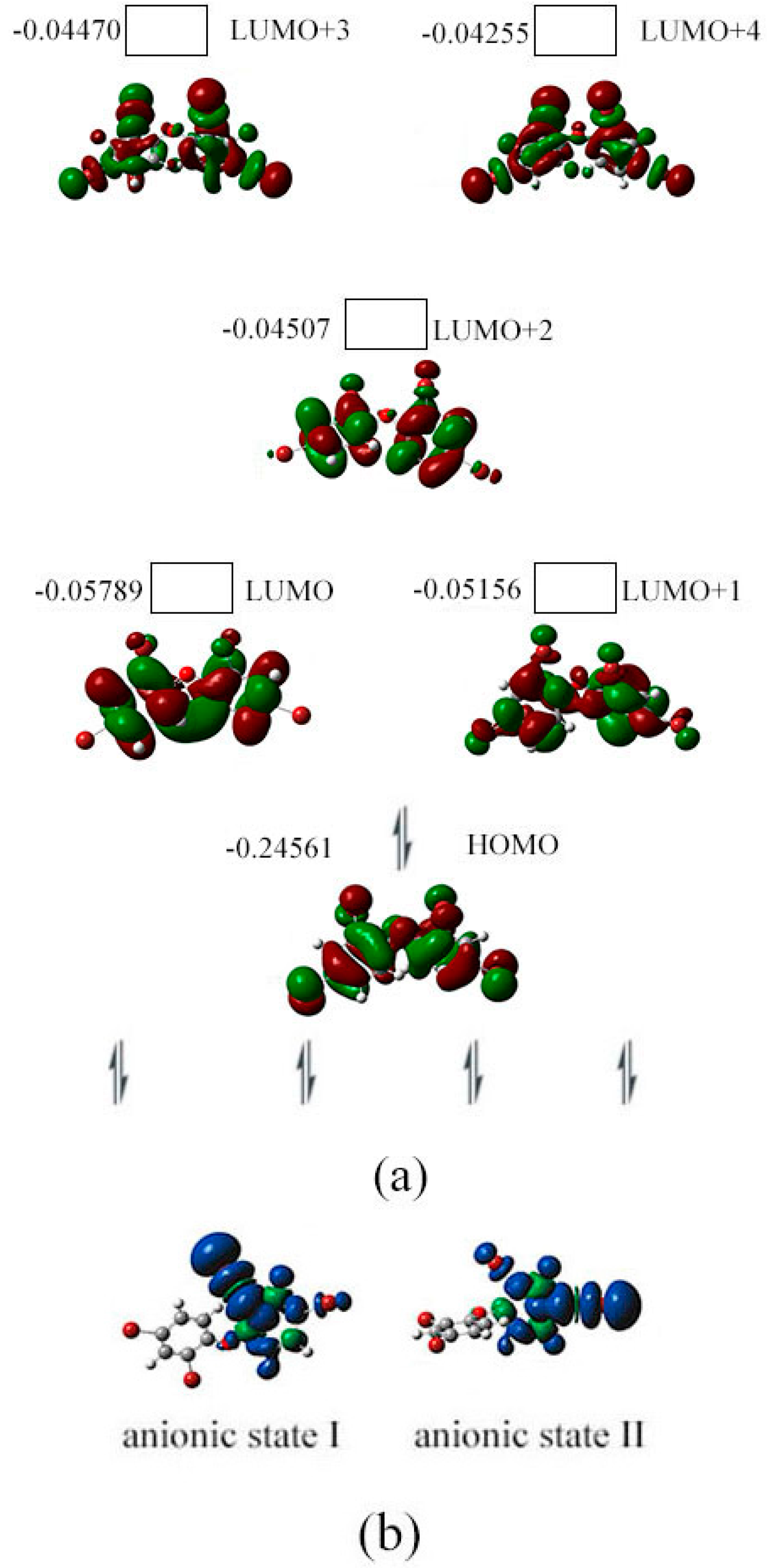
2.3. The Possible Anionic States and the Orbital Energies for the Selected BDE Congeners
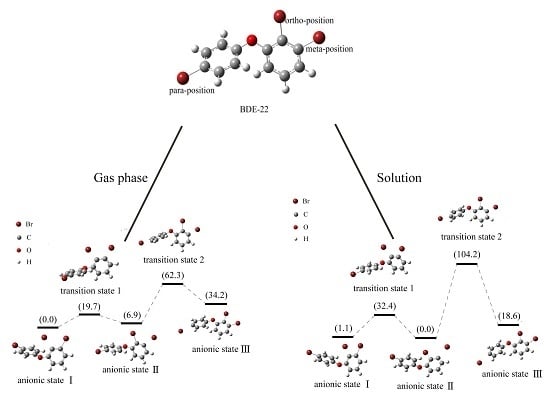
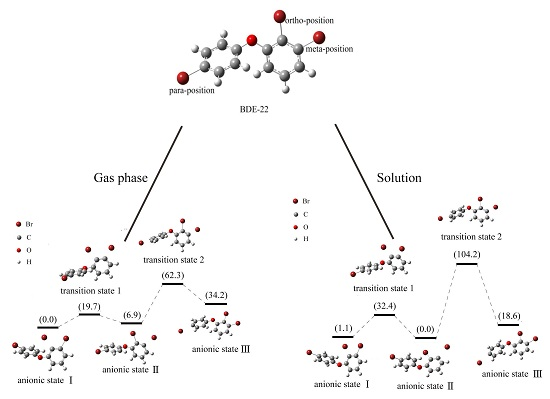
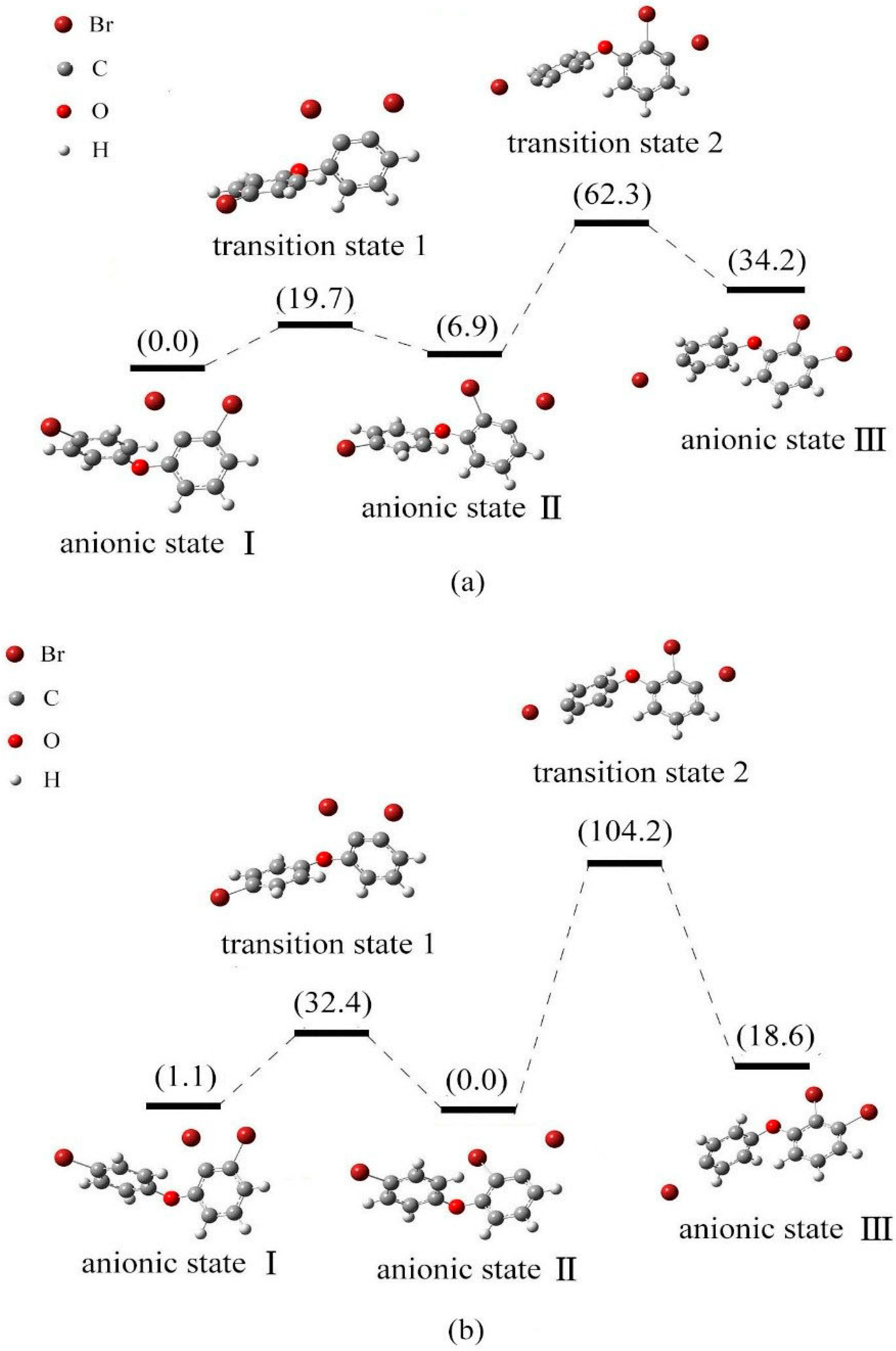
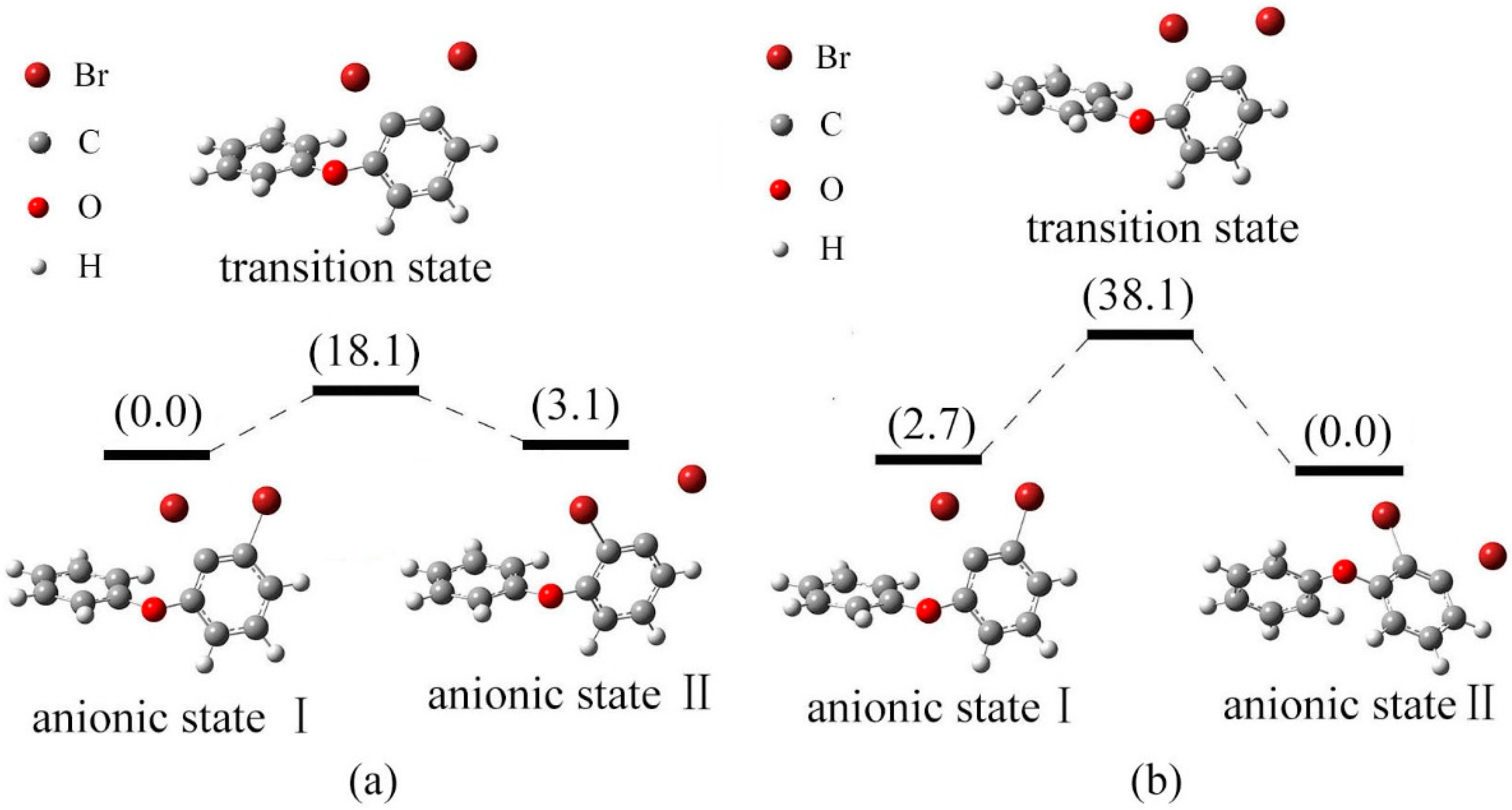
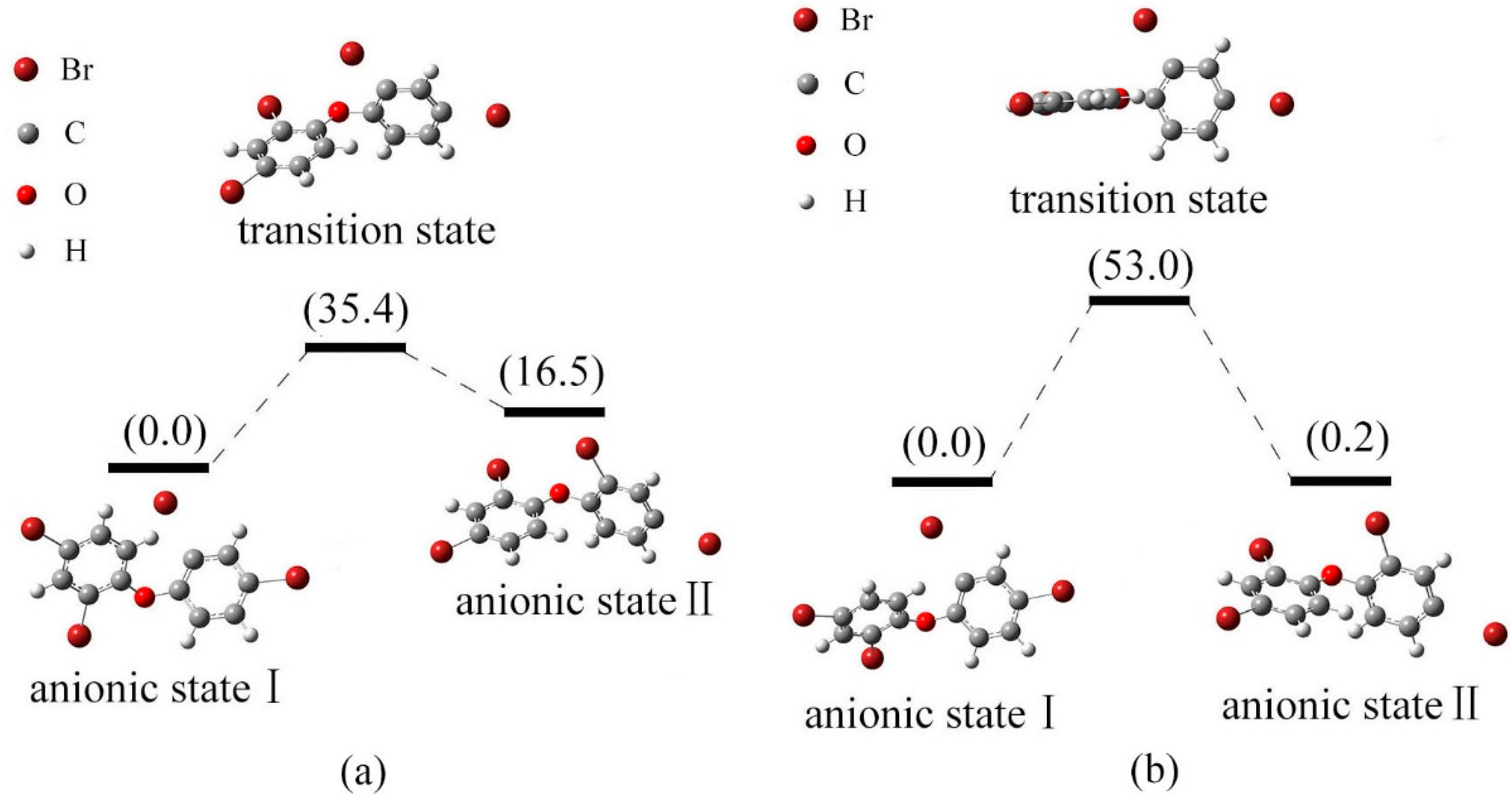
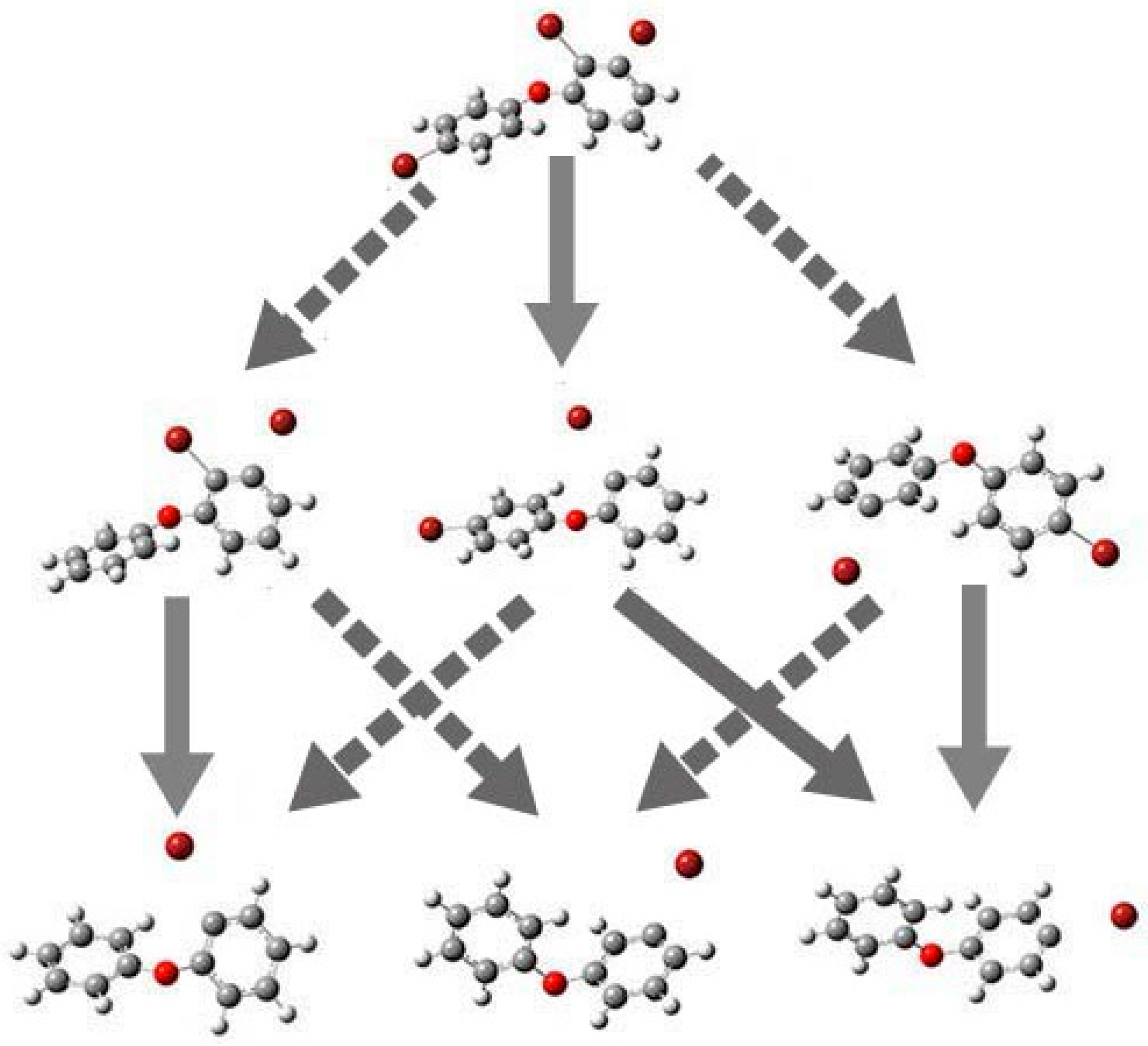
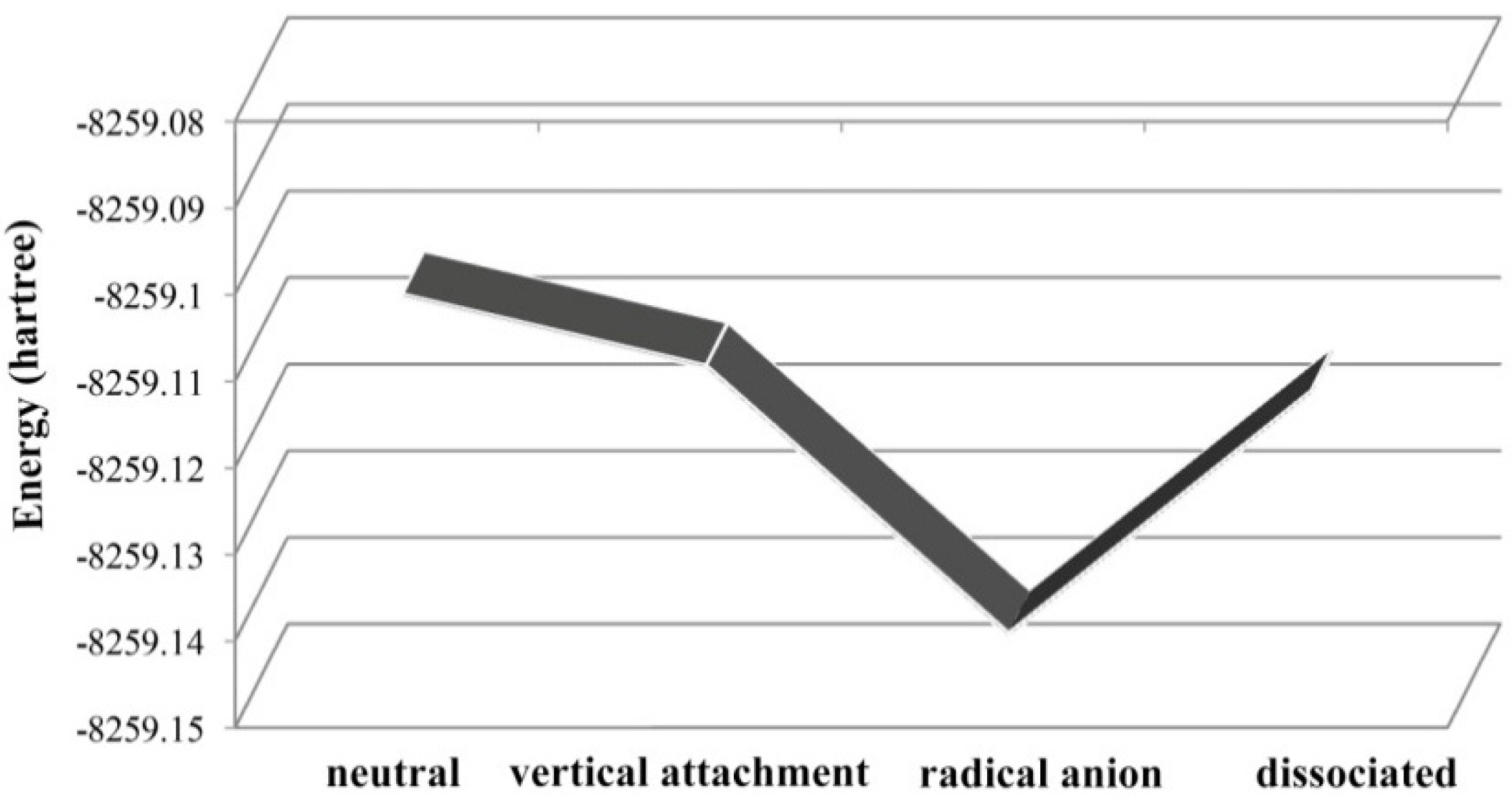
2.4. Use of Optimized Geometries of BDE Congeners to Predict the Dominant Debromimation Pathway
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PBDEs | Polybrominated diphenyl ethers |
| RoHs | The Restriction of the use of Certain Hazardous Substances in Electrical and Electronic Equipment |
| PBB | Polybrominated biphenyls |
| DFT | Density functional theory |
| PM3 | Parametrized model 3 |
| AM1 | Austin model 1 |
| MNDO | Modified neglect of differential overlap |
| TCDD | Tetrachlorodibenzodioxin |
| PCDD | Polychlorinated dibenzo-p-dioxins |
| PCDFs | Polychlorinated dibenzofurans |
| B3LYP | Becke-Lee-Yang-Parr hybrid method |
| CPCM | Conductor-like polarizable continuum model |
| PES | potential energy surface scan |
| TS | transition state |
| IRC | intrinsic reaction coordinated |
| EA | Electron affinities |
| EAAda | Adiabatic electron affinities |
| EAver | Vertical electron affinities |
| ZPC | Zero-point corrections |
| LUMO | The lowest unoccupied molecular orbital |
| HOMO | The highest occupied molecular orbital |
| RMSE | Root mean square error |
References
- Alaee, M.; Arias, P.; Sjödin, A.; Bergman, Å. An overview of commercially used brominated flame retardants, their applications, their use patterns in different countries/regions and possible modes of release. Environ. Int. 2003, 29, 683–689. [Google Scholar] [CrossRef]
- Wang, D.L.; Cai, Z.W.; Jiang, G.B.; Leung, A.; Wong, M.H.; Kwok, W. Determination of polybrominated diphenyl ethers in soil and sediment from an electronic waste recycling facility. Chemosphere 2005, 60, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.J.; Wang, Y.W.; Zhang, A.Q.; Zhang, Q.H.; Zhao, Z.S.; Wang, T.; Jiang, G.B. Spatial distribution of polybrominated biphenyls (PCBs) and polybrominated biphenyl ethers (PBDEs) in an e-waste dismantling region in southeast China: Use of apple snail (Ampullariidae) as a bioindicator. Chemosphere 2011, 82, 648–655. [Google Scholar] [CrossRef] [PubMed]
- He, M.J.; Luo, X.J.; Chen, M.Y.; Sun, Y.X.; Chen, S.J.; Mai, B.X. Bioaccumulation of polybrominated diphenyl ethers and decabromodiphenyl ether in fish from a river system in a highly industrialized area, South China. Sci. Total Environ. 2012, 419, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.M.; Ramos, A.B.A.; Cabrini, T.M.B.; dos Santos Fernandez, M.A. The occurrence of polybrominated diphenyl ethers in Brazil: a review. Int. J. Environ. Health 2015, 7, 247–266. [Google Scholar] [CrossRef]
- Wag̈er, P.A.; Schluep, M.; Müller, E.; Gloor, R. RoHS regulated substances in mixed plastics from waste electrical and electronic equipment. Environ. Sci. Technol. 2012, 46, 628–635. [Google Scholar] [CrossRef] [PubMed]
- He, J.Z.; Robrock Kristin, R.; Alvare-Cohen, L. Microbial reductive debromination of polybrominated diphenyl ethers (PBDEs). Environ. Sci. Technol. 2006, 40, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Huang, J.; Yu, G.; Wang, L.N. Photochemical degradation of six polybrominated diphenyl ether congeners under ultraviolet irradiation in hexane. Chemosphere 2008, 71, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Lution, J.; Luthy Richard, G. Kinetics and pathways for the debromnination of polybrominated diphenyl ethers by bimetallic and nanoscale zerovalent iron: Effects of particle properties and catalyst. Chemosphere 2012, 89, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Tai, C.; Zhao, Z.S.; Wang, Y.W.; Zhang, Q.H.; Jiang, G.B.; Hu, J.T. Debromination of decabrominated diphenyl ether by resin-bond iron nanoparticles. Environ. Sci. Technol. 2007, 41, 6841–6846. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.G.; Zhou, Q.X. Health and ecosystem risks of graphene. Chem. Rev. 2013, 113, 3815–3835. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Freeman, P.K.; Vasil’ev, Y.V.; Voinov, V.G.; Simonich, S.L.; Barofsky, D.F. Theoretical calculation of thermodynamic properties of polybrominated diphenyl ethers. J. Chem. Eng. Data 2005, 50, 1548–1556. [Google Scholar] [CrossRef]
- Hu, J.W.; Kolehmainen, E.; Knuutinen, J. 1H and 13C NMR spectroscopy of brominated diphenyl ethers. A multiple linear regression analysis. Magn. Reson. Chem. 2000, 38, 375–378. [Google Scholar] [CrossRef]
- Eriksson, L.; Hu, J.W. 2,4,6-Tribromodiphenylether. Acta Cryst. E. 2001, 57, 930–932. [Google Scholar] [CrossRef]
- Eriksson, J.; Eriksson, L.; Hu, J.W. 4-Bromophenyl 2,6-dibromophenyl ether. Acta Cryst. E 2002, 58. [Google Scholar] [CrossRef]
- Eriksson, L.; Hu, J.W. 4-Bromophenyl 2,3,4,5,6-pentabromophenyl phenyl ether. Acta Cryst. E 2002, 58, 1147–1149. [Google Scholar] [CrossRef]
- Eriksson, L.; Hu, J.W. 2,3,4,5,6-Pentabromophenyl phenyl ether. Acta Cryst. E 2002, 58, 794–796. [Google Scholar] [CrossRef]
- Eriksson, L.; Hu, J.W. 4-Bromophenyl 2,4-dibromophenyl ether. Acta Cryst. E 2002, 58, 696–698. [Google Scholar] [CrossRef]
- Eriksson, J.; Eriksson, L.; Hu, J.W. 2,4-Dibromophenyl 2, 6-dibromophenyl ether. Acta Cryst. E 2002, 58, 347–349. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef] [PubMed]
- Borisov, Y.; Garrett, B.C.; Mazunov, V.; Nekrasov, Y.S. DFT calculations for the structure and properties of polychlorodibenzo-para-dioxine anion-radicals. J. Struct. Chem. 2005, 46, 591–595. [Google Scholar] [CrossRef]
- Hu, J.W.; Zhuang, Y.; Luo, J.; Wei, X.H.; Huang, X.F. A Theoretical study on reductive debromination of polybrominated diphenyl ethers. Int. J. Mol. Sci. 2012, 13, 9332–9342. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Lin, Y.M.; Hu, J.W. A study on pathway and QSPR models for debromination of PBDEs with pseudopoential method. Adv. Mater. Res. 2014, 997, 25–32. [Google Scholar] [CrossRef]
- Luo, J.; Hu, J.W.; Wei, X.H.; Li, L.Y.; Huang, X.F. Excited states and photodebromination of selected polybrominated diphenyl ethers: Computational and quantitative structure-property relationship studies. Int. J. Mol. Sci. 2015, 16, 1160–1178. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Hu, J.W.; Wei, X.H.; Fu, L.Y.; Li, L.Y. Dehalogenation of persistent halogenated organic compounds: A review of computational studies and quantitative structure-property relationships. Chemosphere 2015, 131, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Foresman, J.B.; Frisch, E. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian, Inc.: Wallingford, CT, USA, 1996. [Google Scholar]
- Hu, J.; Eriksson, L.; Bergman, Å.; Kolehmainen, E.; Knuutinen, J.; Suontamo, R.; Wei, X. Molecular orbital studies on brominated diphenyl ethers. Part I—conformational properties. Chemosphere 2005, 59, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Lynam, M.M.; Kuty, M.; Damborsky, J.; Koca, J.; Adriaens, P. Molecular orbital calculations to describe microbial reductive dechlorination of polychlorinated dioxins. Environ. Toxicol. Chem. 1998, 17, 988–997. [Google Scholar] [CrossRef]
- Pershina, V. Electronic structure and properties of superheavy elements. Nucl. Phys. A 2015, 944, 578–613. [Google Scholar] [CrossRef]
- Yong, D.C. Using existing basis sets. In Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons, Inc.: New York, NY, USA, 2001; Volume 10, pp. 78–98. [Google Scholar]
- Chan, W.T.; Fournier, R. Binding of ammonia to small copper and silver clusters. Chem. Phys. Lett. 1999, 315, 257–265. [Google Scholar] [CrossRef]
- Valdés, Á.; Prosmiti, R.; Villarreal, P.; Delgado-Barrio, G. HeBr2 complex: Ground-state potential and vibrational dynamics from ab initio calculations. Mol. Phys. 2004, 102, 2277–2283. [Google Scholar] [CrossRef]
- Lei, M.; Wang, N.; Zhu, L.H.; Tang, H.Q. Peculiar and rapid photocatalytic degradation of tetrabromodiphenyl ethers over Ag/TiO2 induced by interaction between silver nanoparticles and bromine atoms in the target. Chemosphere 2016, 150, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Li, N.; King, B.R.; Schaefer, H.F., III. Metal triangles versus metal chains and terminal versus bridging hydrogen atoms in trinuclear osmium carbonyl hydride chemistry. New J. Chem. 2014, 38, 1433–1440. [Google Scholar] [CrossRef]
- Sieffert, N.; Bühl, M. Hydrogen generation from alcohols catalyzed by ruthenium−triphenylphosphine complexes: multiple reaction pathways. J. Am. Chem. Soc. 2010, 132, 8056–8070. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Bian, W.S. Theoretical study on the photodegradation mechanism of nona-BDEs in methanol. Chem. Phys. 2013, 14, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Zhang, J.X.; Bian, W.S. Theoretical study on the photodegradation reacton of deca-BDE in THF in the presence of furan. Theor. Chem. Acc. 2016, 135, 4. [Google Scholar] [CrossRef]
- Li, L.Y.; Lin, Y.M.; Hu, J.W. A QSPR study on debromination of PBDEs with CPCM solvation model. Adv. Mater. Res. 2014, 1010–1012, 3–9. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Tao, F.M.; Zeng, E.Y. Structures, reductive dechlorination, and electron affinities of selected polychlorinated dibenzo-p-dioxins: density functional theory study. J. Phys. Chem. A. 2007, 111, 11638–11644. [Google Scholar] [CrossRef] [PubMed]
- Pshenichnyuk, S.A.; Lomakin, G.S.; Modelli, A. Degradation of gas phase decabromodiphenyl ether by resonant interaction with low-energy electrons. Phys. Chem. 2011, 13, 9293–9300. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Ahn, S.; Luthy, R.G. Debromination of polybrominated diphenyl ethers by nanoscale zerovalent iron: pathways, kinetics, and reactivity. Environ. Sci. Technol. 2010, 44, 8236–8242. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Tao, F.M.; Zeng, E.Y. Theoretical study on the chemical properties of polybrominated diphenyl ethers. Chemosphere 2008, 70, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Arulmozhiraja, S.; Morita, M. Electron affinities and reductive dechlorination of toxic polychlorinated dibenzofurans: A density functional theory study. J. Phys. Chem. A 2004, 108, 3499–3508. [Google Scholar] [CrossRef]
- Eloranta, J.; Hu, J.W.; Suontamo, R.; Kolehmainen, E.; Knuutinen, J. Ab initio study of halogenated diphenyl ethers. NMR chemical shift prediction. Magn. Reson. Chem. 2000, 38, 987–993. [Google Scholar] [CrossRef]
- Luo, J.; Hu, J.; Zhuang, Y.; Wei, X.; Huang, X. Electron-induced reductive debromination of 2,3,4-tribromodiphenyl ether: A computational study. J. Mol. Model. 2013, 19, 3333–3338. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM salvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Anderson, M.P.; Uvdal, P. New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple- ζ basis set 6–311+G(d,p). J. Phys. Chem. A 2005, 109, 2937–2941. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, M.V.; Olerich, G. Differentiation of polycyclic aromatic hydrocarbons using electron capture negative chemical ionization. Org. Mass Spectrom. 1984, 19, 486–489. [Google Scholar] [CrossRef]
- Peng, C.; Schlegel, H.B. Combining synchronous transit and Quasi-Newton methods to find transition states. Israel J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comp. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Montgomery, T.; Vreven, K.N.; Kudin, J.C.; Burant, J.M.; Millam, S.S.; et al. Gaussian 03; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Cázares-Larios, U.E.; Reyes-Leaño, U.G.; Castillo-López, P.A.; Pineda-Urbina, K.; Gómez-Sandoval, Z. Computational study of the structure, bonding and reactivity of selected helical metallocenes. Inorg. Chim. Acta 2015, 438, 203–207. [Google Scholar] [CrossRef]
- Lenthe, E.V.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-electron scalar relativistic basis sets for the Lanthanides. J. Chem. Theor. Comput. 2009, 5, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-electron scalar relativistic basis sets for the Actinides. J. Chem. Theor. Comput. 2011, 7, 677–684. [Google Scholar] [CrossRef]
- Williams, S.D.; Edwards, E.E. Scalar relativistic study of the structure of rhodium acetate. Int. J. Mol. Sci. 2004, 5, 67–74. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | IUPAC Name |
|---|---|
| BDE-166 | 2,3,4,4′,5,6-hexa-bromodiphenyl ether |
| BDE-116 | 2,3,4,5,6-penta-bromodiphenyl ether |
| BDE-51 | 2,2′,4,6′-tetra-bromodiphenyl ether |
| BDE-47 | 2,2′,4,4′-tetrabromodiphenyl ether |
| BDE-32 | 2,4′,6-tri-bromodiphenyl ether |
| BDE-30 | 2,4,6-tri-bromodiphenyl ether |
| BDE-28 | 2,4,4′-tri-bromodiphenyl ether |
| BDE-22 | 2,3,4′-tri-bromodiphenyl ether |
| BDE-13 | 3,4′-di-bromodiphenyl ether |
| BDE-8 | 2,4′-di-bromodiphenyl ether |
| BDE-5 | 2,3-di-bromodiphenyl ether |
| BDE-3 | 4-mono-bromodiphenyl ether |
| BDE-2 | 3-mono-bromodiphenyl ether |
| BDE-1 | 2-mono-bromodiphenyl ether |
| Congener | Bond Length | Exptl. (Å) | Reference a | B3LYP/6-311+G(d) | Genecp | ||
|---|---|---|---|---|---|---|---|
| Calculated Value | Error | Calculated Value | Error | ||||
| BDE-28 | Br1–C4 | 1.901 | [18] | 1.917 | 0.016 | 1.945 | 0.044 |
| Br2–C8 | 1.889 | 1.907 | 0.018 | 1.933 | 0.044 | ||
| Br3–C10 | 1.889 | 1.915 | 0.026 | 1.942 | 0.053 | ||
| BDE-30 | Br1–C2 | 1.884 | [14] | 1.905 | 0.021 | 1.931 | 0.047 |
| Br2–C6 | 1.894 | 1.912 | 0.018 | 1.931 | 0.037 | ||
| Br3–C4 | 1.907 | 1.905 | -0.002 | 1.939 | 0.032 | ||
| BDE-32 | Br1–C12 | 1.890 | [15] | 1.908 | 0.018 | 1.947 | 0.057 |
| Br2–C8 | 1.897 | 1.908 | 0.011 | 1.934 | 0.037 | ||
| Br3–C4 | 1.907 | 1.918 | 0.011 | 1.934 | 0.027 | ||
| BDE-51 | Br1–C10 | 1.895 | [19] | 1.915 | 0.020 | 1.943 | 0.048 |
| Br2–C12 | 1.902 | 1.906 | 0.004 | 1.932 | 0.030 | ||
| Br3–C6 | 1.885 | 1.907 | 0.022 | 1.933 | 0.048 | ||
| Br4–C2 | 1.879 | 1.907 | 0.028 | 1.933 | 0.054 | ||
| BDE-116 | Br1–C2 | 1.866 | [17] | 1.899 | 0.033 | 1.925 | 0.059 |
| Br2–C3 | 1.900 | 1.901 | 0.001 | 1.928 | 0.028 | ||
| Br3–C4 | 1.880 | 1.903 | 0.023 | 1.930 | 0.050 | ||
| Br4–C5 | 1.882 | 1.901 | 0.019 | 1.928 | 0.046 | ||
| Br5–C6 | 1.876 | 1.899 | 0.023 | 1.925 | 0.049 | ||
| BDE-166 | Br1–C4 | 1.844 | [16] | 1.917 | 0.073 | 1.945 | 0.101 |
| Br2–C8 | 1.878 | 1.899 | 0.021 | 1.925 | 0.047 | ||
| Br3–C9 | 1.890 | 1.900 | 0.010 | 1.927 | 0.037 | ||
| Br4–C10 | 1.865 | 1.902 | 0.037 | 1.929 | 0.064 | ||
| Br5–C11 | 1.883 | 1.900 | 0.017 | 1.927 | 0.044 | ||
| Br6–C12 | 1.845 | 1.899 | 0.054 | 1.925 | 0.080 | ||
| RMSE | 0.0268 | 0.0511 | |||||
| Congener | Bond Length | Exptl. (Å) | Reference a | B3LYP/6-311+G(d) | Genecp | ||
|---|---|---|---|---|---|---|---|
| Calculated Value | Error | Calculated Value | Error | ||||
| BDE-28 | O–C7 | 1.380 | [18] | 1.371 | −0.009 | 1.370 | −0.010 |
| O–C1 | 1.385 | 1.383 | −0.002 | 1.383 | −0.002 | ||
| BDE-30 | O–C1 | 1.381 | [14] | 1.365 | −0.016 | 1.364 | −0.017 |
| O–C7 | 1.394 | 1.391 | −0.003 | 1.364 | −0.030 | ||
| BDE-32 | O–C7 | 1.384 | [15] | 1.369 | −0.015 | 1.368 | −0.016 |
| O–C1 | 1.388 | 1.385 | −0.003 | 1.385 | −0.003 | ||
| BDE-51 | O–C7 | 1.395 | [19] | 1.375 | −0.020 | 1.374 | −0.021 |
| O–C1 | 1.397 | 1.371 | −0.026 | 1.370 | −0.027 | ||
| BDE-116 | O–C1 | 1.371 | [17] | 1.363 | −0.008 | 1.362 | −0.009 |
| O–C7 | 1.403 | 1.393 | −0.010 | 1.393 | −0.010 | ||
| BDE-166 | O–C7 | 1.391 | [16] | 1.365 | −0.026 | 1.364 | −0.027 |
| O–C1 | 1.414 | 1.389 | −0.025 | 1.389 | −0.025 | ||
| RMSE | 0.0161 | 0.0189 | |||||
| Bond Length | BDE-22 | BDE-13 | BDE-8 | BDE-5 | BDE-3 | BDE-2 | BDE-1 |
|---|---|---|---|---|---|---|---|
| R1 | 1.901 | 1.082 | 1.910 | 1.902 | 1.084 | 1.082 | 1.911 |
| R2 | 1.909 | 1.919 | 1.083 | 1.910 | 1.085 | 1.918 | 1.084 |
| R3 | 1.083 | 1.083 | 1.084 | 1.082 | 1.085 | 1.083 | 1.084 |
| R4 | 1.085 | 1.085 | 1.085 | 1.085 | 1.085 | 1.085 | 1.085 |
| R5 | 1.083 | 1.084 | 1.084 | 1.083 | 1.084 | 1.083 | 1.084 |
| R6 | 1.084 | 1.084 | 1.084 | 1.084 | 1.083 | 1.084 | 1.084 |
| R7 | 1.083 | 1.083 | 1.083 | 1.085 | 1.083 | 1.085 | 1.085 |
| R8 | 1.917 | 1.917 | 1.918 | 1.085 | 1.918 | 1.085 | 1.085 |
| R9 | 1.083 | 1.083 | 1.083 | 1.085 | 1.083 | 1.085 | 1.085 |
| R10 | 1.084 | 1.084 | 1.084 | 1.084 | 1.084 | 1.084 | 1.084 |
| r1 | 1.399 | 1.391 | 1.393 | 1.398 | 1.394 | 1.389 | 1.392 |
| r2 | 1.394 | 1.391 | 1.392 | 1.395 | 1.394 | 1.392 | 1.393 |
| r3 | 1.390 | 1.395 | 1.394 | 1.390 | 1.395 | 1.394 | 1.394 |
| r4 | 1.390 | 1.391 | 1.391 | 1.390 | 1.393 | 1.393 | 1.391 |
| r5 | 1.394 | 1.394 | 1.397 | 1.394 | 1.395 | 1.395 | 1.397 |
| r6 | 1.405 | 1.396 | 1.398 | 1.407 | 1.392 | 1.396 | 1.400 |
| r7 | 1.371 | 1.379 | 1.374 | 1.368 | 1.386 | 1.376 | 1.371 |
| r8 | 1.384 | 1.382 | 1.381 | 1.389 | 1.377 | 1.386 | 1.386 |
| r9 | 1.392 | 1.392 | 1.393 | 1.391 | 1.395 | 1.392 | 1.392 |
| r10 | 1.393 | 1.393 | 1.392 | 1.394 | 1.391 | 1.394 | 1.394 |
| r11 | 1.392 | 1.392 | 1.392 | 1.394 | 1.392 | 1.394 | 1.394 |
| r12 | 1.393 | 1.392 | 1.392 | 1.395 | 1.391 | 1.395 | 1.395 |
| r13 | 1.393 | 1.393 | 1.393 | 1.393 | 1.394 | 1.393 | 1.393 |
| r14 | 1.394 | 1.395 | 1.394 | 1.394 | 1.395 | 1.395 | 1.394 |
| θ1 | 119.0 | 118.4 | 120.4 | 119.1 | 119.5 | 118.8 | 120.5 |
| θ2 | 120.7 | 122.0 | 120.0 | 120.8 | 120.4 | 121.8 | 120.0 |
| θ3 | 119.6 | 118.4 | 119.8 | 119.4 | 119.6 | 118.3 | 119.7 |
| θ4 | 120.6 | 121.0 | 120.2 | 120.7 | 120.6 | 121.4 | 120.3 |
| θ5 | 119.8 | 119.3 | 120.3 | 119.9 | 119.2 | 119.0 | 120.3 |
| θ6 | 120.3 | 120.9 | 119.3 | 120.1 | 120.8 | 120.7 | 119.1 |
| θ7 | 119.8 | 119.9 | 119.9 | 119.3 | 120.1 | 119.4 | 119.4 |
| θ8 | 120.6 | 120.5 | 120.5 | 121.1 | 120.2 | 120.8 | 120.9 |
| θ9 | 119.6 | 119.7 | 119.7 | 119.1 | 119.7 | 119.2 | 119.2 |
| θ10 | 119.6 | 119.6 | 119.7 | 120.5 | 119.7 | 120.5 | 120.6 |
| θ11 | 120.9 | 120.9 | 120.9 | 119.7 | 120.8 | 119.6 | 119.6 |
| θ12 | 119.4 | 119.4 | 119.4 | 120.3 | 119.5 | 120.4 | 120.4 |
| θ13 | 120.7 | 121.0 | 120.7 | 120.7 | 120.8 | 120.8 | 120.8 |
| Bond Length | BDE-22 | BDE-13 | BDE-8 | BDE-5 | BDE-3 | BDE-2 | BDE-1 |
|---|---|---|---|---|---|---|---|
| R1 | 1.958 | 1.088 | 2.793 | 1.957 | 1.085 | 1.087 | 2.806 |
| R2 | 2.635 | 2.754 | 1.086 | 2.668 | 1.086 | 2.786 | 1.086 |
| R3 | 1.087 | 1.087 | 1.088 | 1.087 | 1.085 | 1.087 | 1.088 |
| R4 | 1.088 | 1.089 | 1.087 | 1.088 | 1.086 | 1.089 | 1.087 |
| R5 | 1.085 | 1.085 | 1.087 | 1.085 | 1.083 | 1.085 | 1.087 |
| R6 | 1.083 | 1.083 | 1.085 | 1.083 | 1.088 | 1.085 | 1.085 |
| R7 | 1.084 | 1.084 | 1.084 | 1.083 | 1.088 | 1.086 | 1.087 |
| R8 | 1.928 | 1.928 | 1.933 | 1.085 | 2.780 | 1.085 | 1.086 |
| R9 | 1.084 | 1.084 | 1.083 | 1.086 | 1.088 | 1.086 | 1.086 |
| R10 | 1.084 | 1.085 | 1.081 | 1.085 | 1.088 | 1.083 | 1.081 |
| r1 | 1.368 | 1.387 | 1.388 | 1.368 | 1.390 | 1.386 | 1.387 |
| r2 | 1.389 | 1.388 | 1.399 | 1.388 | 1.397 | 1.387 | 1.399 |
| r3 | 1.397 | 1.399 | 1.397 | 1.397 | 1.393 | 1.399 | 1.397 |
| r4 | 1.396 | 1.398 | 1.393 | 1.396 | 1.395 | 1.398 | 1.393 |
| r5 | 1.392 | 1.391 | 1.401 | 1.393 | 1.399 | 1.392 | 1.401 |
| r6 | 1.400 | 1.395 | 1.382 | 1.400 | 1.401 | 1.396 | 1.383 |
| r7 | 1.400 | 1.411 | 1.403 | 1.397 | 1.365 | 1.408 | 1.400 |
| r8 | 1.364 | 1.361 | 1.365 | 1.368 | 1.409 | 1.365 | 1.369 |
| r9 | 1.399 | 1.401 | 1.397 | 1.399 | 1.392 | 1.398 | 1.396 |
| r10 | 1.390 | 1.390 | 1.395 | 1.390 | 1.402 | 1.395 | 1.394 |
| r11 | 1.392 | 1.392 | 1.389 | 1.397 | 1.386 | 1.394 | 1.394 |
| r12 | 1.389 | 1.389 | 1.391 | 1.393 | 1.386 | 1.397 | 1.396 |
| r13 | 1.394 | 1.394 | 1.390 | 1.394 | 1.402 | 1.390 | 1.391 |
| r14 | 1.397 | 1.399 | 1.401 | 1.397 | 1.392 | 1.401 | 1.401 |
| θ1 | 120.4 | 118.5 | 119.6 | 120.5 | 120.1 | 118.5 | 119.7 |
| θ2 | 120.4 | 121.2 | 120.5 | 120.5 | 120.6 | 121.4 | 120.5 |
| θ3 | 119.9 | 119.6 | 119.7 | 119.7 | 119.0 | 119.5 | 119.7 |
| θ4 | 119.9 | 120.3 | 119.8 | 119.9 | 121.1 | 120.3 | 119.8 |
| θ5 | 119.6 | 118.7 | 119.7 | 119.7 | 119.5 | 118.8 | 119.8 |
| θ6 | 119.8 | 121.8 | 120.7 | 119.6 | 119.7 | 121.6 | 120.5 |
| θ7 | 120.5 | 120.6 | 119.6 | 120.0 | 119.0 | 119.4 | 119.1 |
| θ8 | 119.7 | 119.5 | 119.7 | 119.9 | 121.3 | 119.8 | 119.9 |
| θ9 | 119.9 | 119.9 | 120.7 | 119.4 | 119.0 | 120.1 | 120.3 |
| θ10 | 119.8 | 119.9 | 118.9 | 121.1 | 119.9 | 120.6 | 120.3 |
| θ11 | 120.9 | 120.8 | 121.0 | 119.0 | 120.7 | 119.0 | 119.0 |
| θ12 | 119.3 | 119.3 | 120.0 | 120.6 | 119.9 | 121.2 | 121.4 |
| θ13 | 119.6 | 119.7 | 123.3 | 119.8 | 119.4 | 119.9 | 123.1 |
| Congeners | Ortho-Position | Meta-Position | Para-Position |
|---|---|---|---|
| BDE-22 | 178.5 | −165.7 | 179.9 |
| BDE-13 | - | −162.5 | 179.9 |
| BDE-8 | −174.1 | - | 179.5 |
| BDE-5 | 179.4 | −175.8 | - |
| BDE-3 | - | - | −173.2 |
| BDE-2 | - | 166.5 | - |
| BDE-1 | −177.7 | - | - |
| EA | BDE-22 | BDE-13 | BDE-8 | BDE-5 | BDE-3 | BDE-2 | BDE-1 | |
|---|---|---|---|---|---|---|---|---|
| EAAda | au | 0.0392 | 0.0277 | 0.0306 | 0.0350 | 0.0230 | 0.0238 | 0.0250 |
| eV | 1.066 | 0.753 | 0.832 | 0.951 | 0.626 | 0.647 | 0.681 | |
| EAver | au | 0.0082 | 0.0046 | 0.0029 | 0.0017 | −0.0053 | −0.0061 | −0.0080 |
| eV | 0.223 | 0.124 | 0.079 | 0.046 | −0.145 | −0.167 | −0.218 | |
| Orbit | BDE-47 | BDE-22 | BDE-13 | BDE-8 | BDE-5 | BDE-3 | BDE-2 | BDE-1 |
|---|---|---|---|---|---|---|---|---|
| EHOMO | −0.24561 | −0.24321 | −0.24051 | −0.23786 | −0.2419 | −0.23279 | −0.23812 | −0.23537 |
| ELUMO | −0.05789 | −0.05047 | −0.04592 | −0.0443 | −0.04342 | −0.03813 | −0.03744 | −0.03597 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Hu, J.; Shi, X.; Ruan, W.; Luo, J.; Wei, X. Theoretical Studies on Structures, Properties and Dominant Debromination Pathways for Selected Polybrominated Diphenyl Ethers. Int. J. Mol. Sci. 2016, 17, 927. https://doi.org/10.3390/ijms17060927
Li L, Hu J, Shi X, Ruan W, Luo J, Wei X. Theoretical Studies on Structures, Properties and Dominant Debromination Pathways for Selected Polybrominated Diphenyl Ethers. International Journal of Molecular Sciences. 2016; 17(6):927. https://doi.org/10.3390/ijms17060927
Chicago/Turabian StyleLi, Lingyun, Jiwei Hu, Xuedan Shi, Wenqian Ruan, Jin Luo, and Xionghui Wei. 2016. "Theoretical Studies on Structures, Properties and Dominant Debromination Pathways for Selected Polybrominated Diphenyl Ethers" International Journal of Molecular Sciences 17, no. 6: 927. https://doi.org/10.3390/ijms17060927