
Targeting the Innate Immune Response to Improve Cardiac Graft Recovery after Heart Transplantation: Implications for the Donation after Cardiac Death

Abstract
:
1. Introduction
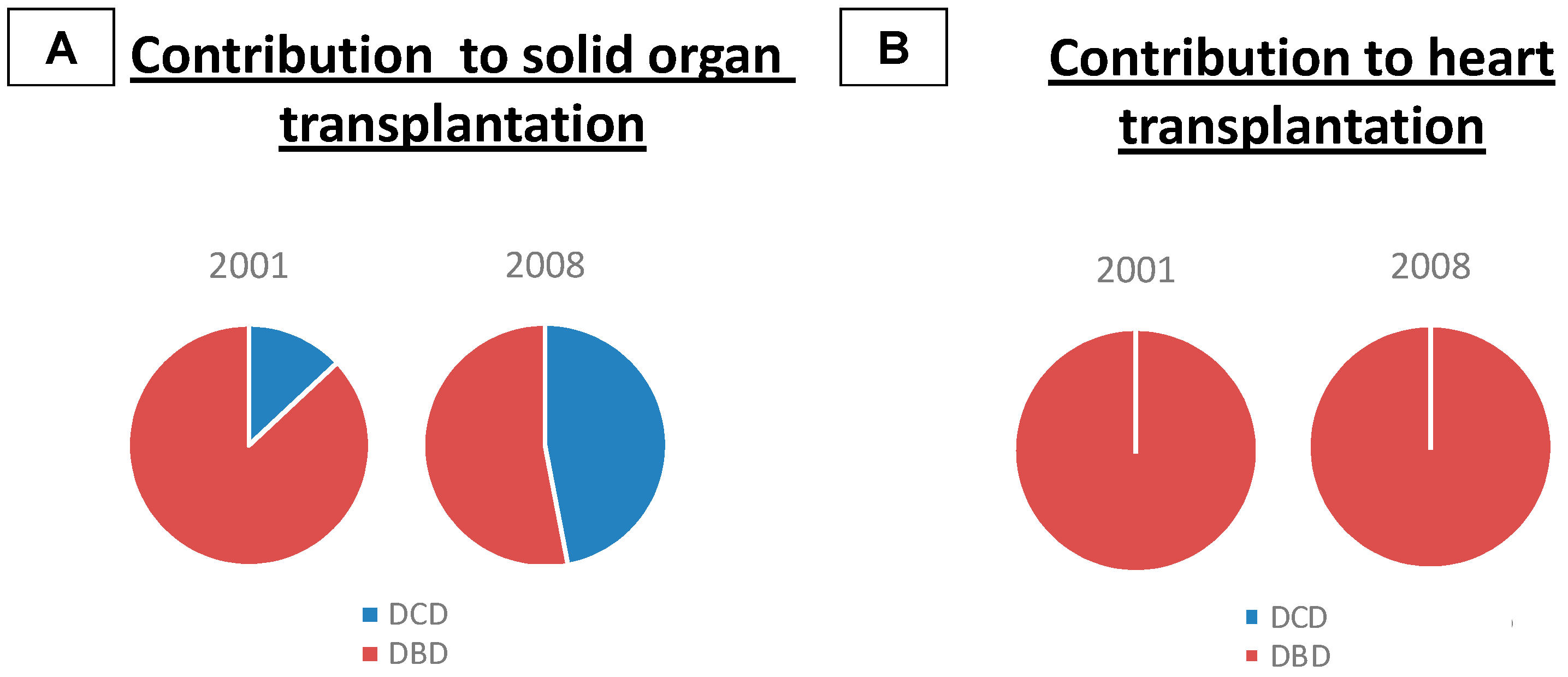
2. Types of Donors
Heart Transplantation from Donors after Cardiac Death
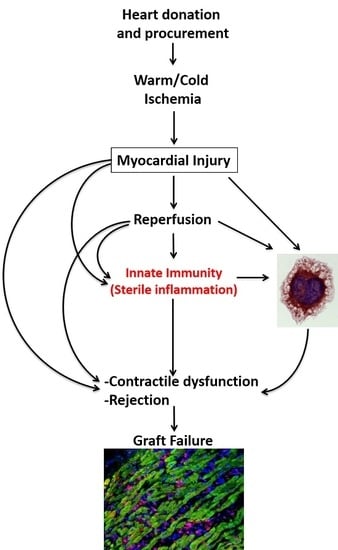
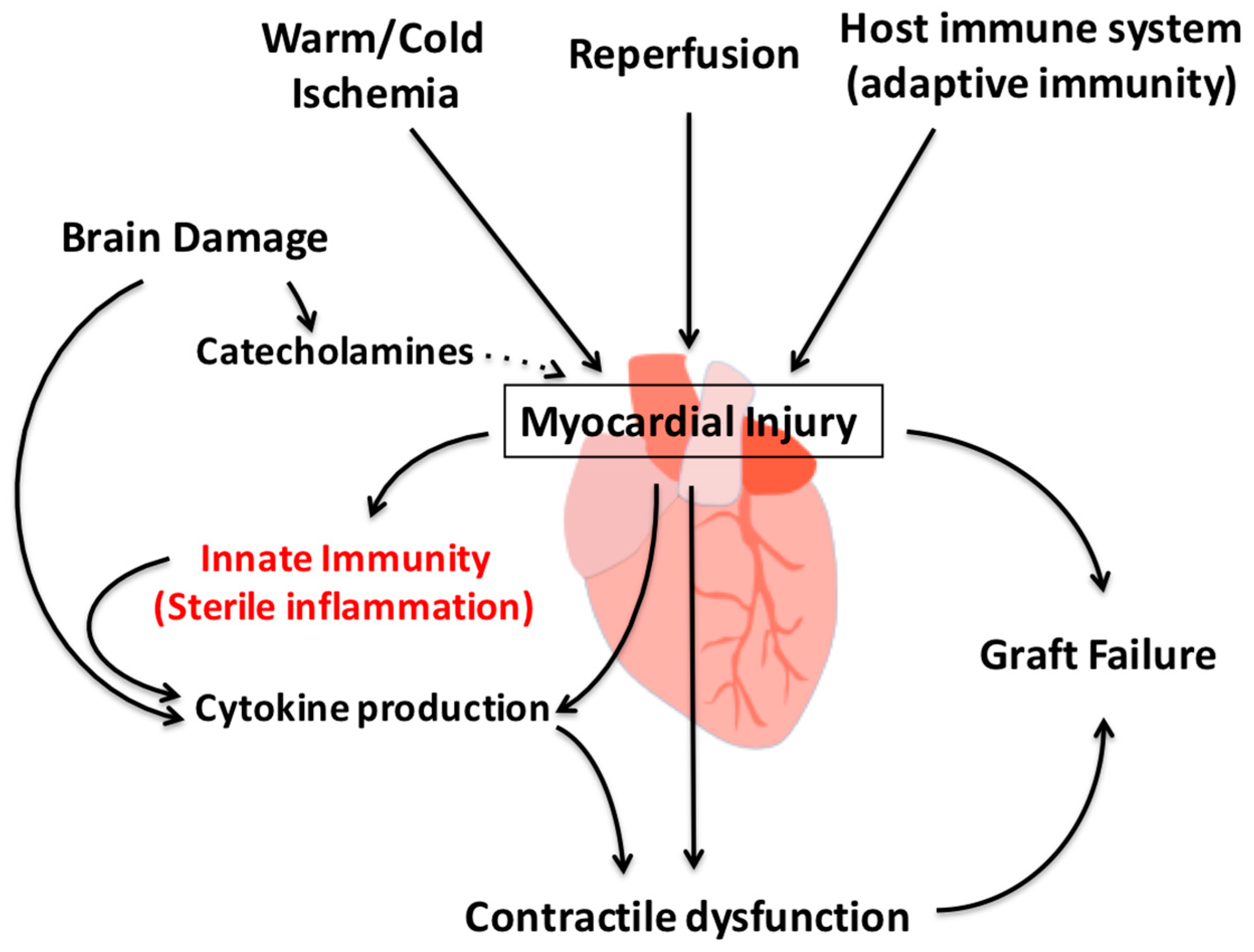
3. Myocardial Injury during Organ Procurement
4. Innate Immune Response during Organ Procurement
4.1. The Inflammasome
4.2. Inflammatory Injury in the DBD Heart
4.3. Inflammation during Cold Ischemia
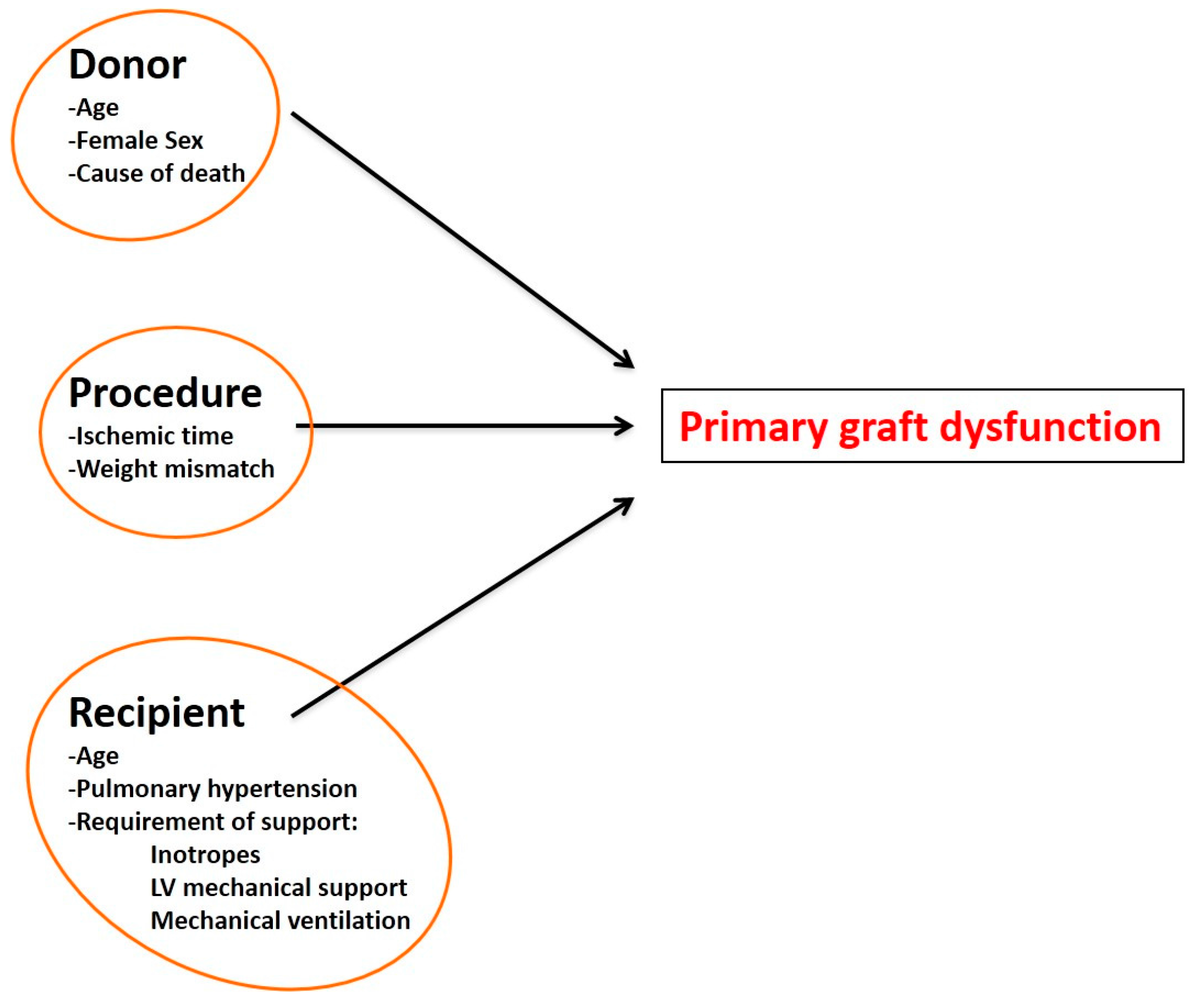
5. Primary Graft Dysfunction after Transplantation
6. Immune Response Leading to Acute or Late Rejection
6.1. Hyperacute and Acute Rejection
6.2. Late Rejection
7. Immune Response following Transplantation
7.1. Lymphocytic Response
7.2. Innate Immunity and Myocardial Injury following Transplantation
8. The Innate Immune Response as a Potential Pharmacological Target
8.1. Interleukin-1α and Its Role as Alarmin
8.2. Targeting the Toll-Like Receptors (TLRs) Pathway
8.3. Targeting the NLRP3 Inflammasome Pathway
8.4. Interleukin-1 Blockade
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mancini, D.; Lietz, K. Selection of cardiac transplantation candidates in 2010. Circulation 2010, 122, 173–183. [Google Scholar] [CrossRef] [PubMed]
- American Heart Association. Available online: http://www.heart.org/HEARTORG/Conditions/HeartFailure/Advanced-Heart-Failure_UCM_441925_Article.jsp (accessed on 2 March 2016).
- Benden, C.; Goldfarb, S.B.; Edwards, L.B.; Kucheryavaya, A.Y.; Christie, J.D.; Dipchand, A.I.; Dobbels, F.; Levvey, B.J.; Lund, L.H.; Meiser, B.; et al. The registry of the International Society for Heart and Lung Transplantation: Seventeenth official pediatric lung and heart-lung transplantation report—2014; Focus theme: Retransplantation. J. Heart Lung Transpl. 2014, 33, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 Update: A report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed]
- Roger, V.L. Epidemiology of heart failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. The war against heart failure: The Lancet lecture. Lancet 2015, 385, 812–824. [Google Scholar] [CrossRef]
- Smolina, K.; Wright, F.L.; Rayner, M.; Goldacre, M.J. Determinants of the decline in mortality from acute myocardial infarction in England between 2002 and 2010: Linked national database study. BMJ 2012, 344, d8059. [Google Scholar] [CrossRef] [PubMed]
- Colvin-Adams, M.; Smithy, J.M.; Heubner, B.M.; Skeans, M.A.; Edwards, L.B.; Waller, C.; Schnitzler, M.A.; Snyder, J.J.; Israni, A.K.; Kasiske, B.L. OPTN/SRTR 2012 Annual Data Report: Heart. Am. J. Transpl. 2014, 14, 113–138. [Google Scholar] [CrossRef] [PubMed]
- Neyrinck, A.; van Raemdonck, D.; Monbaliu, D. Donation after circulatory death: Current status. Curr. Opin. Anaesthesiol. 2013, 26, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, S.; Mandrekar, P. Cellular stress response and innate immune signaling: Integrating pathways in host defense and inflammation. J. Leukoc. Biol. 2013, 94, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Saidi, R.F.; Bradley, J.; Greer, D.; Luskin, R.; O’Connor, K.; Delmonico, F.; Kennealey, P.; Pathan, F.; Schuetz, C.; Elias, N.; et al. Changing pattern of organ donation at a single center: Are potential brain dead donors being lost to donation after cardiac death? Am. J. Transpl. 2010, 10, 2536–2540. [Google Scholar] [CrossRef]
- Laks, H.; Scholl, F.G.; Drinkwater, D.C.; Blitz, A.; Hamilton, M.; Moriguchi, J.; Fonarow, G.; Kobashigawa, J. The alternate recipient list for heart transplantation: Does it work? J. Heart Lung Transpl. 1997, 16, 735–742. [Google Scholar]
- Chen, J.M.; Russo, M.J.; Hammond, K.M.; Mancini, D.M.; Kherani, A.R.; Fal, J.M.; Mazzeo, P.A.; Pinney, S.P.; Edwards, N.M.; Naka, Y. Alternate waiting list strategies for heart transplantation maximize donor organ utilization. Ann. Thorac. Surg. 2005, 80, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Laks, H.; Marelli, D.; Fonarow, G.C.; Hamilton, M.A.; Ardehali, A.; Moriguchi, J.D.; Bresson, J.; Gjertson, D.; Kobashigawa, J.A.; UCLA Heart Tranplant Group. Use of two recipient lists for adults requiring heart transplantation. J. Thorac. Cardiovasc. Surg. 2003, 125, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Poston, R.S.; Griffith, B.P. Heart transplantation. J. Intensive Care Med. 2004, 19, 3–12. [Google Scholar] [CrossRef] [PubMed]
- López-Navidad, A.; Caballero, F. Extended criteria for organ acceptance. Strategies for achieving organ safety and for increasing organ pool. Clin. Transpl. 2003, 17, 308–324. [Google Scholar] [CrossRef]
- Wittwer, T.; Wahlers, T. Marginal donor grafts in heart transplantation: Lessons learned from 25 years of experience. Transpl. Int. 2008, 2, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Orioles, A.; Morrison, W.E.; Rossano, J.W.; Shore, P.M.; Hasz, R.D.; Martiner, A.C.; Berg, R.A.; Nadkarni, V.M. An under-recognized benefit of cardiopulmonary resuscitation: Organ transplantation. Crit. Care Med. 2013, 41, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Quader, M.A.; Wolfe, L.G.; Kasirajan, V. Heart transplantation outcomes from cardiac arrest-resuscitated donors. J. Heart Lung Transpl. 2013, 32, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Quader, M.; Wolfe, L.; Katlaps, G.; Kasirajan, V. Donor heart utilization following cardiopulmonary arrest and resuscitation: Influence of donor characteristics and wait times in transplant regions. J. Transpl. 2014, 2014, 519401. [Google Scholar] [CrossRef] [PubMed]
- Blackstock, M.J.; Ray, D.C. Organ donation after circulatory death: An update. Eur. J. Emerg. Med. 2014, 21, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, P.E.; Monaco, A.P. Donation after circulatory death: Current practices, ongoing challenges, and potential improvements. Transplantation 2014, 97, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Pomfret, E.A.; Sung, R.S.; Allan, J.; Kinkhabwala, M.; Melancon, J.K.; Roberts, J.P. Solving the organ shortage crisis: The 7th annual American Society of Transplant Surgeons’ State-of-the-Art Winter Symposium. Am. J. Transpl. 2008, 8, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Osaki, S.; Locher, M.R.; Lushaj, E.B.; Akhter, S.A.; Kohmoto, T. Functional evaluation of human donation after cardiac death donor hearts using a continuous isolated myocardial perfusion technique: Potential for expansion of the cardiac donor population. J. Thorac. Cardiovasc. Surg. 2014, 148, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, M.; Dornbierer, M.; Clément, D.; Gahl, B.; Dick, F.; Carrel, T.P.; Tevaearai, H.T.; Longnus, S. Mild hypothermia during global cardiac ischemia opens a window of opportunity to develop heart donation after cardiac death. Transpl. Int. 2013, 26, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Gries, C.J.; White, D.B.; Truog, R.D.; Dubois, J.; Cosio, C.C.; Dhanani, S.; Chan, K.M.; Corris, P.; Dark, J.; Fulda, G.; et al. An official American Thoracic Society/International Society for Heart and Lung Transplantation/Society of Critical Care Medicine/Association of Organ and Procurement Organizations/United Network of Organ Sharing Statement: Ethical and policy considerations in organ donation after circulatory determination of death. Am. J. Respir. Crit. Care Med. 2013, 188, 103–109. [Google Scholar] [PubMed]
- Dalle Ave, A.L.; Shaw, D.M.; Pascual, M.; Benaroyo, L. Heart donation after circulatory determination of death: Ethically acceptable? Nat. Rev. Cardiol. 2014, 11, 553. [Google Scholar] [CrossRef] [PubMed]
- Osaki, S.; Anderson, J.E.; Johnson, M.R.; Edwards, N.M.; Kohmoto, T. The potential of cardiac allografts from donors after cardiac death at the University of Wisconsin Organ Procurement Organization. Eur. J. Cardiothorac. Surg. 2010, 37, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Noterdaeme, T.; Detry, O.; Hans, M.F.; Nellessen, E.; Ledoux, D.; Joris, J.; Meurisse, M.; Defraigne, J.O. What is the potential increase in the heart graft pool by cardiac donation after circulatory death? Transpl. Int. 2013, 26, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Birati, E.Y.; Rame, J.E. Left ventricular assist device management and complications. Crit. Care Clin. 2014, 30, 607–627. [Google Scholar] [CrossRef] [PubMed]
- Mancini, D.; Colombo, P.C. Left ventricular assist devices: A rapidly evolving alternative to transplant. J. Am. Coll. Cardiol. 2015, 65, 2542–2555. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, L.W.; Rose, E.A. Left ventricular assist devices: Bridges to transplantation, recovery, and destination for whom? Circulation 2003, 108, 3059–3063. [Google Scholar] [CrossRef] [PubMed]
- Jaski, B.E.; Kim, J.C.; Naftel, D.C.; Jarcho, J.; Costanzo, M.R.; Eisen, H.J.; Kirklin, J.K.; Bourge, R.C.; Cardiac Transplant Research Database Research Group. Cardiac transplant outcome of patients supported on left ventricular assist device vs. intravenous inotropic therapy. J. Heart Lung Transpl. 2001, 20, 449–456. [Google Scholar] [CrossRef]
- Dhital, K.K.; Iyer, A.; Connellan, M.; Chew, H.C.; Gao, L.; Doyle, A.; Hicks, M.; Kumarasinghe, G.; Soto, C.; Dinale, A.; et al. Adult heart transplantation with distant procurement and ex vivo preservation of donor hearts after circulatory death: A case series. Lancet 2015, 385, 2585–2591. [Google Scholar] [CrossRef]
- McKeown, D.W.; Bonser, R.S.; Kellum, J.A. Management of the heartbeating brain-dead organ donor. Br. J. Anaesth. 2012, 108, i96–i107. [Google Scholar] [CrossRef] [PubMed]
- Mackersie, R.; Bronsther, O.; Shackford, S. Organ procurement in patients with fatal head injuries. The fate of the potential donor. Ann. Surg. 1991, 213, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Bugge, J. Brain death and its implications for management of the potential organ donor. Acta Anaesthesiol. Scand. 2009, 53, 1239–1250. [Google Scholar] [CrossRef] [PubMed]
- Smith, M. Physiologic changes during brain stem death—Lessons for management of the organ donor. J. Heart Lung Transpl. 2004, 23, S217–S222. [Google Scholar] [CrossRef] [PubMed]
- Marasco, S.F.; Kras, A.; Schulberg, E.; Vale, M.; Lee, G.A. Impact of warm ischemia time on survival after heart transplantation. Transpl. Proc. 2012, 44, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Mitropoulos, F.A.; Odim, J.; Marelli, D.; Karandikar, K.; Gjertson, D.; Ardehali, A.; Kobashigawa, J.; Laks, H. Outcome of hearts with cold ischemic time greater than 300 min. A case-matched study. Eur. J. Cardiothorac. Surg. 2005, 28, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Guibert, E.E.; Petrenko, A.Y.; Balaban, C.L.; Somov, A.Y.; Rodriguez, J.V.; Fuller, B.J. Organ preservation: Current concepts and new strategies for the next decade. Transfus. Med. Hemother. 2011, 38, 125–142. [Google Scholar] [CrossRef] [PubMed]
- McAnulty, J.F. Hypothermic organ preservation by static storage methods: Current status and a view to the future. Cryobiology 2010, 60, S13–S19. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. The pathogenesis of coronary artery disease and the acute coronary syndromes. N. Engl. J. Med. 1992, 326, 242–250. [Google Scholar] [PubMed]
- Jennings, R.B.; Reimer, K.A.; Steenbergen, C. Myocardial ischemia revisited. The osmolar load, membrane damage, and reperfusion. J. Mol. Cell. Cardiol. 1986, 18, 769–780. [Google Scholar] [CrossRef]
- Fuller, B.; Guibert, E.; Rodriguez, J. Lessons from natural cold-induced dormancy to organ preservation in medicine and biotechnology: From the ‘backwoods to the bedside’. In Dormancy and Resistance to Harsh Environments; Lubens, E., Cerda, J., Clark, M., Eds.; Topics in Current Genetics; Springer-Verlag Berlin Heidelberg: Berlin, Germany, 2010; pp. 253–278. [Google Scholar]
- Rauen, U.; de Groot, H. New insights into the cellular and molecular mechanisms of cold storage injury. J. Investig. Med. 2004, 52, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Hosgood, S.A.; Bagul, A.; Nicholson, M.L. Minimising cold ischaemic injury in an experimental model of kidney transplantation. Eur. J. Clin. Investig. 2010, 41, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Del Rizzo, D.F.; Menkis, A.H.; Pflugfelder, P.W.; Novick, R.J.; McKenzie, F.N.; Boyd, W.D.; Kostuk, W.J. The role of donor age and ischemic time on survival following orthotopic heart transplantation. J. Heart Lung Transpl. 1999, 18, 310–319. [Google Scholar] [CrossRef]
- Southard, J.; Belzer, F.O. Organ preservation. Annu. Rev. Med. 1995, 46, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Desrois, M.; Piccardo, A.; Zogheib, E.; Dalmasso, C.; Lan, C.; Fourré, D.; Cozzone, P.J.; Caus, T.; Bernard, M. Heart donation after cardiac death: Preliminary study on an isolated, perfused swine heart after 20 min of normothermic ischemia. Transpl. Proc. 2014, 46, 3314–3318. [Google Scholar] [CrossRef] [PubMed]
- Van Caenegem, O.; Beauloye, C.; Bertrand, L.; Horman, S.; Lepropre, S.; Sparavier, G.; Vercruysse, J.; Bethuyne, N.; Poncelet, A.J.; Gianello, P.; et al. Hypothermic continuous machine perfusion enables preservation of energy charge and functional recovery of heart grafts in an ex vivo model of donation following circulatory death. Eur. J. Cardiothorac. Surg. 2015. [Google Scholar] [CrossRef]
- Niemann, C.U.; Feiner, J.; Swain, S.; Bunting, S.; Friedman, M.; Crutchfield, M.; Broglio, K.; Hirose, R.; Roberts, J.P.; Malinoski, D. Therapeutic hypothermia in deceased organ donors and kidney-graft function. N. Engl. J. Med. 2015, 373, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Hicks, M.; Hing, A.; Gao, L.; Ryan, J.; Macdonald, P.S. Organ preservation. Methods Mol. Biol. 2006, 333, 331–374. [Google Scholar] [PubMed]
- Barry, W. Mechanisms of myocardial cell injury during ischemia and reperfusion. J. Card. Surg. 1987, 2, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Seccombe, J.F.; Schaff, H.V. Coronary artery endothelial function after myocardial ischemia and reperfusion. Ann. Thorac. Surg. 1995, 60, 778–788. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Fedak, P.W.; Weisel, R.D.; Butany, J.; Rao, V.; Maitland, A.; Li, R.K.; Dhillon, B.; Yau, T.M. Fundamentals of reperfusion injury for the clinical cardiologist. Circulation 2002, 105, 2332–2336. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Burleson, K.O.; Kloner, R.A.; Babior, B.M.; Engler, R.L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Investig. 1994, 94, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Marchant, D.J.; Boyd, J.H.; Lin, D.C.; Granville, D.J.; Garmaroudi, F.S.; McManus, B.M. Inflammation in myocardial diseases. Circ. Res. 2012, 110, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.A.; White, P.; Xiang, B.; Lin, H.Y.; Tsui, S.S.; Ashley, E.; Lee, T.W.; Klein, J.R.; Kumar, K.; Arora, R.C.; et al. Hearts from DCD donors display acceptable biventricular function after heart transplantation in pigs. Am. J. Transpl. 2011, 11, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Neumar, R.W.; Nolan, J.P.; Adrie, C.; Aibiki, M.; Berg, R.A.; Böttiger, B.W.; Callaway, C.; Clark, R.S.; Geocadin, R.G.; Jauch, E.C.; et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 2008, 118, 2452–2483. [Google Scholar] [PubMed]
- Iyer, A.; Kumarasinghe, G.; Hicks, M.; Watson, A.; Gao, L.; Doyle, A.; Keogh, A.; Kotlyar, E.; Hayward, C.; Dhital, K.; et al. Primary graft failure after heart transplantation. J. Transpl. 2011, 2011, 175768. [Google Scholar] [CrossRef] [PubMed]
- Zipes, D.P.; Wellens, H.J. Sudden cardiac death. Circulation 1998, 98, 2334–2351. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Taylor, D.O. Advances in the understanding and management of heart transplantation. F1000Prime Rep. 2015, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Kreisel, D.; Goldstein, D.R. Processes of Sterile Inflammation. J. Immunol. 2013, 191, 2857–2863. [Google Scholar] [CrossRef] [PubMed]
- LaRosa, D.F.; Rahman, A.H.; Turka, L.A. The innate immune system in allograft rejection and tolerance. J. Immunol. 2007, 178, 7503–7509. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; de Kleijn, D.P.; Pasterkamp, G. Innate immune signaling in cardiac ischemia. Nat. Rev. Cardiol. 2011, 8, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; Mauro, A.G.; Salloum, F.; van Tassell, B.W.; Abbate, A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid. Redox Signal. 2015, 22, 1146–1161. [Google Scholar] [CrossRef] [PubMed]
- Zedler, S.; Faist, E. The impact of endogenous triggers on trauma-associated inflammation. Curr. Opin. Crit. Care 2006, 12, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Monack, D.M. Inflammasome adaptors and sensors: Intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 2007, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.; Opitz, B.; van der Meer, J.H.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Toldo, S.; Mezzaroma, E.; Abbate, A. Targeting interleukin-1 in heart disease. Circulation 2013, 128, 1910–1923. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome Activation of Cardiac Fibroblasts Is Essential for Myocardial Ischemia/Reperfusion Injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef] [PubMed]
- Sandanger, Ø.; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Bracey, N.A.; Beck, P.L.; Muruve, D.A.; Hirota, S.A.; Guo, J.; Jabagi, H.; Wright, J.R., Jr.; Macdonald, J.A.; Lees-Miller, J.P.; Roach, D.; et al. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp. Physiol. 2013, 98, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Kannan, H.; Bussani, R.; Anzini, M.; Sonnino, C.; Sinagra, G.; Merlo, M.; Mezzaroma, E.; De-Giorgio, F.; Silvestri, F.; et al. Formation of the inflammasome in acute myocarditis. Int. J. Cardiol. 2014, 171, e119–e121. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; Abbate, A. Interleukin-1 Blockade in Acute Myocardial Infarction and Heart Failure: Ready for Clinical Translation? Transl. Med. 2012, 3, e114. [Google Scholar]
- Lund, L.H.; Edwards, L.B.; Kucheryavaya, A.Y.; Dipchand, A.I.; Benden, C.; Christie, J.D.; Dobbels, F.; Kirk, R.; Rahmel, A.O.; Yusen, R.D.; et al. The Registry of the International Society for Heart and Lung Transplantation: Thirtieth official adult heart transplant report—2013; Focus theme: Age. J. Heart Lung Transpl. 2013, 32, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.R.; Dipchand, A.; Starling, R.; Anderson, A.; Chan, M.; Desai, S.; Fedson, S.; Fisher, P.; Gonzales-Stawinski, G.; Martinelli, L.; et al. The International Society of Heart and Lung Transplantation Guidelines for the care of heart transplant recipients. J. Heart Lung Transpl. 2010, 29, 914–956. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.P.; Bittner, H.B.; Kendall, S.W.; van Trigt, P. Hormonal and hemodynamic changes in a validated animal model of brain death. Crit. Care Med. 1996, 24, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C.; Varela, J.C.; Tomlinson, S. Complement-dependent inflammation and injury in a murine model of brain dead donor hearts. Circ. Res. 2009, 105, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C.; Floerchinger, B.; Qiao, F.; Casey, S.; Williamson, T.; Moseley, E.; Stoica, S.; Goddard, M.; Ge, X.; Tullius, S.G.; et al. Donor brain death exacerbates complement-dependent ischemia/reperfusion injury in transplanted hearts. Circulation 2013, 127, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Danobeitia, J.S.; Djamali, A.; Fernandez, L.A. The role of complement in the pathogenesis of renal ischemia-reperfusion injury and fibrosis. Fibrogenes. Tissue Repair 2014, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Damman, J.; Hoeger, S.; Boneschansker, L.; Theruvath, A.; Waldherr, R.; Leuvenink, H.G.; Ploeg, R.J.; Yard, B.A.; Seelen, M.A. Targeting complement activation in brain-dead donors improves renal function after transplantation. Transpl. Immunol. 2011, 24, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Floerchinger, B.; Yuan, X.; Jurisch, A.; Timsit, M.O.; Ge, X.; Lee, Y.L.; Schmid, C.; Tullius, S.G. Inflammatory immune responses in a reproducible mouse brain death model. Transpl. Immunol. 2012, 27, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.G.; Evans, W.J.; Hughes, V.A.; Meredith, C.N.; Dinarello, C.A. Physiological mechanisms contributing to increased interleukin-1 secretion. J. Appl. Physiol. 1986, 61, 1869–1874. [Google Scholar] [PubMed]
- Van der Poll, T.; Lowry, S.F. Epinephrine inhibits endotoxin-induced IL-1 beta production: Roles of tumor necrosis factor-alpha and IL-10. Am. J. Physiol. 1997, 273, R1885–R1890. [Google Scholar] [PubMed]
- Takada, M.; Nadeau, K.C.; Shaw, G.D.; Marquette, K.A.; Tilney, N.L. The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. Inhibition by a soluble P-selectin ligand. J. Clin. Investig. 1997, 99, 2682–2690. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, J.; Wang, S.; Liu, K.; Wang, L.; Huang, L. Hmgb1-TLR4-IL-23-IL-17A axis promote ischemia-reperfusion injury in a cardiac transplantation model. Transplantation 2013, 95, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Dare, A.J.; Logan, A.; Prime, T.A.; Rogatti, S.; Goddard, M.; Bolton, E.M.; Bradley, J.A.; Pettigrew, G.J.; Murphy, M.P.; Saeb-Parsy, K. The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J. Heart Lung Transpl. 2015, 34, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Wenceslau, C.F.; McCarthy, C.G.; Szasz, T.; Spitler, K.; Goulopoulou, S.; Webb, R.C.; Working Group on DAMPs in Cardiovascular Disease. Mitochondrial damage-associated molecular patterns and vascular function. Eur. Heart J. 2014, 35, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, M.; Franchin, M.; Soldini, G.; Ietto, G.; Chiappa, C.; Maritan, E.; Villa, F.; Carcano, G.; Dionigi, R. Impact of static cold storage VS hypothermic machine preservation on ischemic kidney graft: Inflammatory cytokines and adhesion molecules as markers of ischemia/reperfusion tissue damage. Our preliminary results. Int. J. Surg. 2013, 11, S110–S114. [Google Scholar] [CrossRef]
- Christia, P.; Frangogiannis, N.G. Targeting inflammatory pathways in myocardial infarction. Eur. J. Clin. Investig. 2013, 43, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Kobashigawa, J.; Zuckermann, A.; Macdonald, P.; Leprince, P.; Esmailian, F.; Luu, M.; Mancini, D.; Patel, J.; Razi, R.; Reichenspurner, H.; et al. Report from a consensus conference on primary graft dysfunction after cardiac transplantation. J. Heart Lung Transpl. 2014, 33, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Chew, H.C.; Kumarasinghe, G.; Iyer, A.; Hicks, M.; Gao, L.; Doyle, A.; Jabbour, A.; Dhital, K.; Granger, E.; Jansz, P.; et al. Primary Graft Dysfunction After Heart Transplantation. Curr. Transpl. Rep. 2014, 1, 257–265. [Google Scholar] [CrossRef]
- O’Brien, L.C.; Mezzaroma, E.; van Tassell, B.W.; Marchetti, C.; Carbone, S.; Abbate, A.; Toldo, S. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol. Med. 2014, 20, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Crudele, V.; Picascia, A.; Infante, T.; Grimaldi, V.; Maiello, C.; Napoli, C. Repeated immune and non immune insults to the graft after heart transplantation. Immunol. Lett. 2011, 141, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Afzali, B.; Lechler, R.I.; Hernandez-Fuentes, M.P. Allorecognition and the allore-sponse: Clinical implications. Tissue Antigens 2007, 69, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Gallon, L.G.; Leventhal, J.R.; Kaufman, D.B. Pretransplantevaluation of renal transplant candidates. Semin. Nephrol. 2002, 22, 515–525. [Google Scholar] [PubMed]
- Stewart, S.; Winters, G.L.; Fishbein, M.C.; Tazelaar, H.D.; Kobashigawa, J.; Abrams, J.; Andersen, C.B.; Angelini, A.; Berry, G.J.; Burke, M.M.; et al. Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J. Heart Lung Transpl. 2005, 24, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Gass, A.L.; Emaminia, A.; Lanier, G.; Aggarwal, C.; Brown, K.A.; Raffa, M.; Kai, M.; Spielvogel, D.; Malekan, R.; Tang, G.; et al. Cardiac Transplantation in the New Era. Cardiol. Rev. 2015, 23, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Chih, S.; Chruscinski, A.; Ross, H.J.; Tinckam, K.; Butany, J.; Rao, V. Antibody-mediated rejection: An evolving entity in heart transplantation. J. Transpl. 2012, 2012, 210210. [Google Scholar] [CrossRef] [PubMed]
- You, S. Differential sensitivity of regulatory and effector T cells to cell death: A prerequisite for transplant tolerance. Front. Immunol. 2015, 6, 242. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.A.; Eid, R.E.; Qin, L.; Yi, T.; Kirkiles-Smith, N.C.; Tellides, G.; Pober, J.S. Interleukin (IL)-1 promotes allogeneic T cell intimal infiltration and IL-17 production in a model of human artery rejection. J. Exp. Med. 2008, 205, 3145–3158. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, D.J.; Nakao, A.; McCurry, K.R.; Billiar, T.R. Toll-like receptors and myocardial ischemia/reperfusion, inflammation, and injury. Curr. Cardiol. 2009, 5, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Vilahur, G.; Badimon, L. Ischemia/reperfusion activates myocardial innate immune response: The key role of the Toll-like receptor. Front. Physiol. 2014, 5, 496. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.J.; Gracon, A.S.; Ripsch, M.S.; Fisher, A.J.; Cheon, B.M.; Pandya, P.H.; Vittal, R.; Capitano, M.L.; Kim, Y.; Allette, Y.M.; et al. The HMGB1-RAGE axis mediates traumatic brain injury-induced pulmonary dysfunction in lung transplantation. Sci. Transl. Med. 2014, 6, 2521–2524. [Google Scholar] [CrossRef] [PubMed]
- Stribos, E.G.; van Werkhoven, M.B.; Poppelaars, F.; van Goor, H.; Olinga, P.; van Son, W.J.; Damman, J.; Seelen, M.A. Renal expression of Toll-like receptor 2 and 4: Dynamics in human allograft injury and comparison to rodents. Mol. Immunol. 2015, 64, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Krüger, B.; Krick, S.; Dhillon, N.; Lerner, S.M.; Ames, S.; Bromberg, J.S.; Lin, M.; Walsh, L.; Vella, J.; Fischereder, M.; et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 93390–93395. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, U.; Bergler, T.; Rihm, M.; Pace, C.; Krüger, B.; Jung, B.; Reinhold, S.W.; Farkas, S.; Rümmele, P.; Krämer, B.K.; et al. Impact of Toll-like receptor 2 expression in renal allograft rejection. Nephrol. Dial. Transpl. 2011, 26, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Beduhn, M.; Zheng, X.; Lian, D.; Chen, D.; Li, R.; Siu, L.K.; Marleau, A.; French, P.W.; Ichim, T.E.; et al. Induction of alloimmune tolerance in heart transplantation through gene silencing of TLR adaptors. Am. J. Transpl. 2012, 12, 2675–2688. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Schmaderer, C.; Kiss, E.; Schmidt, C.; Bonrouhi, M.; Porubsky, S.; Gretz, N.; Schaefer, L.; Kirschning, C.J.; Popovic, Z.V.; et al. Recipient Toll-like receptors contribute to chronic graft dysfunction by both MyD88- and TRIF-dependent signaling. Dis. Model. Mech. 2010, 3, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; McGeough, M.D.; Peña, C.A.; Marchetti, C.; Sonnino, C.; van Tassell, B.W.; Salloum, F.N.; Voelkel, N.F.; Hoffman, H.M.; et al. Independent roles of the priming and the triggering of the NLRP3 inflammasome in the heart. Cardiovasc. Res. 2015, 105, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. NLRP3 Inflammasome as a Novel Player in Myocardial Infarction. Int. Heart J. 2011, 55, 101–105. [Google Scholar] [CrossRef]
- Toldo, S.; Zhong, H.; Mezzaroma, E.; van Tassell, B.W.; Kannan, H.; Zeng, D.; Belardinelli, L.; Voelkel, N.F.; Abbate, A. GS-6201, a selective blocker of the A2B adenosine receptor, attenuates cardiac remodeling after acute myocardial infarction in the mouse. J. Pharmacol. Exp. Ther. 2012, 343, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Xie, M.; Xu, L.; Zheng, X.; Yang, Y.; Lv, X. The protective role of interleukin-18 binding protein in a murine model of cardiac ischemia/reperfusion injury. Transpl. Int. 2015, 28, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, D.; Ganesan, J.; Bscheider, M.; Stickel, N.; Weber, F.C.; Guarda, G.; Follo, M.; Pfeifer, D.; Tardivel, A.; Ludigs, K.; et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J. Exp. Med. 2013, 210, 1899–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, T.; Kamijo, S.; Wada, Y.; Yamaura, K.; Takahashi, K.; Komatsu, K.; Otsu, Y.; Terasaki, T.; Fukui, D.; Amano, J.; et al. Upregulation of the apoptosis-related inflammasome in cardiac allograft rejection. J. Heart Lung Transpl. 2010, 29, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.B.; Mauro, A.G.; Flattery, M.; Toldo, S.; Abbate, A. Formation of the inflammasome during cardiac allograft rejection. Int. J. Cardiol. 2015, 201, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Marasco, S.F.; Sheeran, F.L.; Chaudhuri, K.; Vale, M.; Bailey, M.; Pepe, S. Molecular markers of programmed cell death in donor hearts before transplantation. J. Heart Lung Transpl. 2014, 33, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.G.; Brough, D.; Freeman, S. Inhibiting the Inflammasome: A Chemical Perspective. J. Med. Chem. 2016, 59, 1691–1710. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; van Tassell, B.W.; Zhang, S.; Abbate, A. A Novel Pharmacologic Inhibitor of the NLRP3 Inflammasome Limits Myocardial Injury after Ischemia-Reperfusion in the Mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Toldo, S.; Chojnacki, J.; Mezzaroma, E.; Liu, K.; Salloum, F.N.; Nordio, A.; Carbone, S.; Mauro, A.G.; Das, A.; et al. Pharmacologic Inhibition of the NLRP3 Inflammasome Preserves Cardiac Function After Ischemic and Nonischemic Injury in the Mouse. J. Cardiovasc. Pharmacol. 2015, 66, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.; Stokes, L.; Sluyter, R. The P2X7 receptor channel: Recent developments and the use of P2X7 antagonists in models of disease. Pharmacol. Rev. 2014, 66, 638–675. [Google Scholar] [CrossRef] [PubMed]
- Polosa, R.; Blackburn, M.R. Adenosine receptors as targets for therapeutic intervention in asthma and chronic obstructive pulmonary disease. Trends Pharmacol. Sci. 2009, 30, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Seropian, I.M.; Mezzaroma, E.; van Tassell, B.W.; Salloum, F.N.; Lewis, E.C.; Voelkel, N.; Dinarello, C.A.; Abbate, A. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2011, 51, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mauro, A.G.; Marchetti, C.; Gelber, C.; Mezzaroma, E.; Wolpe, S.; Yachin, G.; Salloum, F.N.; van Tassell, B.; Abbate, A. Anti-inflammatory peptide SP16 reduces infarct size after myocardial ischemia and reperfusion in the mouse. Eur. Heart J. 2015, 36 (Suppl. 1), 1205–1210. [Google Scholar]
- Toldo, S.; Das, A.; Mezzaroma, E.; Chau, V.Q.; Marchetti, C.; Durrant, D.; Samidurai, A.; van Tassell, B.W.; Yin, C.; Ockaili, R.A.; et al. Induction of microRNA-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ. Cardiovasc. Genet. 2014, 7, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Throne, M.L.; Amar, N.J.; Sebai, M.; Kivitz, A.J.; Kavanaugh, A.; Weinstein, S.P.; Belomestnov, P.; Yancopoulos, G.D.; Stahl, N.; et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: Results from two sequential placebo-controlled studies. Arthritis Rheumatol. 2008, 58, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, H.J.; Kone-Paut, I.; Kuemmerle-Deschner, J.B.; Leslie, K.S.; Hachulla, E.; Quartier, P.; Gitton, X.; Widmer, A.; Patel, N.; Hawkins, P.N.; et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med. 2009, 360, 2416–2425. [Google Scholar] [CrossRef] [PubMed]
- Neven, B.; Marvillet, I.; Terrada, C.; Ferster, A.; Boddaert, N.; Couloignier, V.; Pinto, G.; Pagnier, A.; Bodemer, C.; Bodaghi, B.; et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheumatol. 2010, 62, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; van Tassell, B.W.; Farkas, D.; Marchetti, C.; Voelkel, N.F.; Abbate, A. Interleukin-1β blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp. Physiol. 2013, 98, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Schatz, A.M.; Mezzaroma, E.; Chawla, R.; Stallard, T.W.; Stallard, W.C.; Jahangiri, A.; van Tassell, B.W.; Abbate, A. Recombinant human interleukin-1 receptor antagonist provides cardioprotection during myocardial ischemia reperfusion in the mouse. Cardiovasc. Drugs Ther. 2012, 26, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; Bressi, E.; Marchetti, C.; Carbone, S.; Sonnino, C.; van Tassell, B.W.; Abbate, A. Interleukin-1β blockade improves left ventricular systolic/diastolic function and restores contractility reserve in severe ischemic cardiomyopathy in the mouse. J. Cardiovasc. Pharmacol. 2014, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; van Tassell, B.W.; Seropian, I.M.; Toldo, S.; Robati, R.; Varma, A.; Salloum, F.N.; Smithson, L.; Dinarello, C.A. Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur. J. Heart Fail. 2010, 12, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Varma, A.; Salloum, F.N.; Das, A.; Seropian, I.M.; Toldo, S.; Smithson, L.; Hoke, N.N.; Chau, V.Q.; Robati, R.; et al. Interleukin-1 trap attenuates cardiac remodeling after experimental acute myocardial infarction in mice. J. Cardiovasc. Pharmacol. 2010, 55, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Salloum, F.N.; Chau, V.; Varma, A.; Hoke, N.N.; Toldo, S.; Biondi-Zoccai, G.G.; Crea, F.; Vetrovec, G.W.; Abbate, A. Anakinra in experimental acute myocardial infarction—Does dosage or duration of treatment matter? Cardiovasc. Drugs Ther. 2009, 23, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Salloum, F.N.; Vecile, E.; Das, A.; Hoke, N.N.; Straino, S.; Biondi-Zoccai, G.G.; Houser, J.E.; Qureshi, I.Z.; Ownby, E.D.; et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 2008, 117, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; van Tassell, B.W.; Biondi-Zoccai, G.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Biondi-Zoccai, G.G.; van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.A.; Roberts, C.S.; Varma, A.; et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am. J. Cardiol. 2010, 105, 1371.e1–1377.e1. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Arena, R.A.; Toldo, S.; Mezzaroma, E.; Azam, T.; Seropian, I.M.; Shah, K.; Canada, J.; Voelkel, N.F.; Dinarello, C.A.; et al. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS ONE 2012, 7, e33438. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Arena, R.; Biondi-Zoccai, G.; Canada, J.; Oddi, C.; Abouzaki, N.A.; Jahangiri, A.; Falcao, R.A.; Kontos, M.C.; Shah, K.B.; et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am. J. Cardiol. 2014, 113, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Thuren, T.; Zalewski, A.; Libby, P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: Rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am. Heart J. 2011, 162, 597–605. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviations | Full Names |
|---|---|
| AAT | Alpha-1Antitrypsin |
| ACR | Acute cellular rejection |
| AdorA2B | Adenosine Receptor A2B |
| AMI | Acute Myocardial Infarction |
| AMR | Antibody-mediated rejection |
| APC | Antigen Presenting Cells |
| ASC | Apoptosis Speck-Like Protein containing a Caspase recruiting domain (CARD) |
| ATP | Adenosine Triphosphate |
| β-AR | β-Adrenergic Receptor |
| CARD | Caspase recruiting domain |
| CAV | Coronary Artery Vasculopathy |
| DAMPS | Damage Associated Molecular Patterns |
| DBD | Donation after Brain Death |
| DCD | Donation after Cardiac Death |
| HF | Heart Failure |
| HMGB-1 | High-mobility group protein B1 |
| HMP | Hypothermic Machine Perfusion |
| HTx | Heart Transplantation |
| IL-1 | Interleukin-1 |
| IL-18BP | Interleukin-18 Binding Protein |
| IL-1Ra | IL-1 receptor antagonist |
| IL-1RAp | IL-1R Accessory protein |
| IL-1RI | Interleukin-1 receptor type 1 |
| IRFs | Interferon Regulated Transcription Factors |
| ISHLT | International Society of Heart and Lung Transplantation |
| LRRs | Leucine-rich repeats |
| LVADs | Left Ventricular Assisting Devices |
| MAPK | Mitogen Activated Protein Kinases |
| MHC | Major Histocompatibility Complex |
| MyD88 | Myeloid Differentiation Factor 88 |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NF-kB | Nuclear Factor-κB |
| NLRP3 | NOD Like Receptors (NLR) containing a Pyrin Domain |
| PGD | Primary Graft Dysfunction |
| PRRs | Pattern Recognition Receptors |
| P2X7R | Purinergic 2X Receptor 7 |
| PYD | PYRIN Domain |
| ROS | Reactive Oxygen Species |
| SCS | Static Cold Storage |
| TIR | Toll/Interleukin-1 Receptor |
| TIRAP | Toll/Interleukin-1 Receptor (TIR) domain containing an adaptor protein |
| TLR | Toll-Like Receptor |
| TNF-α | Tumor Necrosis Factor-alpha |
| TRAP | Toll/Interleukin-1 Receptor (TIR) Adaptor Protein |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toldo, S.; Quader, M.; Salloum, F.N.; Mezzaroma, E.; Abbate, A. Targeting the Innate Immune Response to Improve Cardiac Graft Recovery after Heart Transplantation: Implications for the Donation after Cardiac Death. Int. J. Mol. Sci. 2016, 17, 958. https://doi.org/10.3390/ijms17060958
Toldo S, Quader M, Salloum FN, Mezzaroma E, Abbate A. Targeting the Innate Immune Response to Improve Cardiac Graft Recovery after Heart Transplantation: Implications for the Donation after Cardiac Death. International Journal of Molecular Sciences. 2016; 17(6):958. https://doi.org/10.3390/ijms17060958
Chicago/Turabian StyleToldo, Stefano, Mohammed Quader, Fadi N. Salloum, Eleonora Mezzaroma, and Antonio Abbate. 2016. "Targeting the Innate Immune Response to Improve Cardiac Graft Recovery after Heart Transplantation: Implications for the Donation after Cardiac Death" International Journal of Molecular Sciences 17, no. 6: 958. https://doi.org/10.3390/ijms17060958